

- •Государственная фармакопея республики беларусь первое издание
- •Республики Беларусь
- •1. Общие сведения
- •1.1. Общие положения
- •1.2. Другие положения, распространяющиеся на общие и частные фармакопейные статьи
- •Условия хранения лекарственного средства
- •Пределы, указываемые на упаковке
- •1.5. Сокращения и обозначения
- •1.6. Единицы международной системы (си), используемые в фармакопейных статьях, и их соответствие другим единицам
- •2. Методы анализа
- •2.1. Оборудование
- •2.1.1. Каплемер
- •2.1.2. Сравнительная таблица пористости стеклянных фильтров
- •Пористость фильтра (ф.Евр.) (1)
- •Максимальный диаметр пор в микрометрах
- •2.1.3. Лампы с ультрафиолетовым излучением для аналитических целей
- •2.1.4. Сита
- •2.2. Физические и физико-химические методы
- •2.2.1. Определение прозрачности и степени мутности жидкостей
- •2.2.2. Определение степени окрашивания жидкостей
- •2.2.3. Потенциометрическое определение рН
- •2.2.4. Зависимость между реакцией раствора, приблизительным значением рН и цветом индикаторов
- •Изменение цвета
- •2.2.5. Относительная плотность
- •2.2.6. Показатель преломления (индекс рефракции)
- •2.2.7. Оптическое вращение
- •2.2.8. Вязкость
- •1/Прив 1
- •2.2.9. Метод капиллярной вискозиметрии
- •2.2.10. Метод ротационной вискозиметрии
- •2.2.11. Температурные пределы перегонки
- •2.2.14. Температура плавления - капиллярный метод
- •2.2.17. Температура каплепадения
- •2.2.18. Температура затвердевания
- •2.2.21. Флуориметрия
- •2.2.22. Атомно-эмиссионная спектрометрия
- •2.2.23. Атомно-абсорбционная спектрометрия
- •2.2.24. Абсорбционная спектрофотометрия в инфракрасной
- •2.2.25. Абсорбционная спектрофотометрия в ультрафиолетовой видимой областях
- •2. Многокомпонентный спектрофотометрический анализ.
- •2.2.26. Бумажная хроматография
- •2.2.27. Тонкослойная хроматография
- •2.2.28. Газовая хроматография
- •2.2.29. Жидкостная хроматография
- •2.2.30. Эксклюзионная хроматография
- •2.2.31. Электрофорез
- •2.2.32. Потеря в массе при высушивании
- •2.2.33. Спектрометрия ядерного магнитного резонанса
- •2.2.34. Термогравиметрия
- •2.2.35. Осмоляльность
- •2.2.36. Потенциометрическое определение концентрации ионов с использованием ионселективных электродов
- •2.2.37. Рентгенофлуоресцентная спектрометрия
- •2.2.38. Удельная электропроводность
- •2.2.39. Молекулярно-массовое распределение декстранов
- •2.2.40. Спектрофотометрия ближнего ик-диапазона
- •2.2.41. Круговой дихроизм
- •2.2.42. Плотность твердых тел
- •2.2.43. Масс-спектрометрия
- •2.2.44. Определение содержания общего органического углерода в воде для фармацевтического применения
- •2.2.45. Сверхкритическая флюидная хроматография
- •2.2.46. Хроматографические методы разделения
- •2.2.47. Капиллярный электрофорез
- •2.2.48. Рамановская спектрометрия (# спектрометрия комбинационного рассеяния)
- •2.2.54. Изоэлектрическое фокусирование
- •2.3.1. Реакции подлинности (идентификации) на ионы и функциональные группы
- •2.3.2. Идентификация жирных масел методом тонкослойной хроматографии
- •2.3.3. Идентификация фенотиазинов методом тонкослойной хроматографии
- •2.3.4. Определение запаха
- •2.4. Испытания на предельное содержание примесей
- •2.4.1. Аммония соли
- •2.4.2. Мышьяк
- •2.4.3. Кальций
- •2.4.6. Магний
- •2.4.7. Магний и щелочноземельные металлы
- •2.4.8. Тяжелые металлы
- •2.4.15. Никель в полиолах
- •2.4.1.6. Общая зола
- •2.4.21. Посторонние масла в жирных маслах методом тонкослойной хроматографии
- •2.4.22. Посторонние жирные кислоты в маслах методом газовой хроматографии
- •2.4.23. Стерины в жирных маслах
- •2.4.24. Идентификация остаточных растворителей и их количественное определение
- •2.4.25. Остаточные количества этиленоксида и диоксана
- •2.4.27. Никель в гидрогенизированных растительных маслах
- •2.5. Методы количественного определения 2.5.1. Кислотное число
- •2.5.3. Гидроксильное число
- •2.5.4. Йодное число
- •2.5.5. Перекисное (пероксидное) число
- •2.5.6. Число омыления
- •2.5.7. Неомыляемые вещества
- •2.5.8. Определение аминного азота в соединениях, которые содержат первичную ароматическую аминогруппу
- •2.5.9. Определение азота после минерализации серной кислотой
- •2.5.10. Метод сжигания в колбе с кислородом
- •2.5.11. Комплексометрическое титрование
- •2.5.12. Вода: полумикрометод (#Метод к.Фишера)
- •2.5.13. Алюминий в адсорбированных вакцинах
- •2.5.14. Кальций в адсорбированных вакцинах
- •2.5.20. Гексозамины в полисахаридных вакцинах
- •2.5.21. Метилпентозы в полисахаридных вакцинах
- •2.5.24. Диоксид углерода в газах
- •2.5.25. Оксид углерода в газах
- •2.5.26. Оксид азота и диоксид азота в газах
- •2.5.27. Кислород в газах
- •2.5.30. Окисляющие вещества
- •2.5.33. Общий белок
- •2.5.34. Уксусная кислота в синтетических пептидах
- •2.6. Биологические испытания
- •2.6.1. Стерильность
- •2.6.2. Микобактерии
- •2.6.3. Испытания на посторонние вирусы с использованием куриных эмбрионов
- •2.6.4. Испытание на вирусы лейкоза
- •2.6.5. Испытание на посторонние вирусы с использованием клеточных культур
- •2.6.6. Испытание на посторонние агенты с использованием цыплят.
- •2.6.7. Микоплазмы
- •2.6.8 Пирогенность
- •2.6.9. Аномальная токсичность
- •2.6.10. Гистамин
- •2.6.11. Депрессорные вещества
- •2.6.12. Микробиологические испытания нестерильной продукции (суммарное количество жизнеспособных аэробов)
- •2.6.13. Микробилогические испытания нестерильной продукции (испытания на наличие специфических микроорганизмов)
- •0,9 % Раствор натрия хлорида
- •1 % Раствор фенолового красного
- •0,5 % Раствор малахитового зеленого
- •2.6.14. Бактериальные эндотоксины
- •1. Предварительные испытания
- •2. Предельное испытание (метод а) (I) Методика
- •2. Полуколичественное испытание (метод в)
- •1. Турбидиметрический принцип (методы с и f)
- •2.6.15. Активатор прекалликреина
- •2.6.16. Испытания на посторонние агенты в вирусных вакцинах для медицинского применения
- •2.6.17. Испытание на антикомплементарную активность иммуноглобулина
- •2.6.18. Испытание живых вирусных вакцин на нейровирулентность
- •2.6.19. Испытание пероральной вакцины полиомиелита на нейровирулентность
- •5.1. Предотвращение загрязнения
- •5.4 Детектирование
- •7.1 Валидация системы для количественного определения методом
- •7.2. Контроль качества реагентов.
- •7.3. Контроль хода испытания.
- •7.4. Внешняя оценка качества
- •2.6.22. Активированные факторы свертывания крови
- •2.7 Биологические методы количественного определения
- •2.7.1. Иммунохимические методы
- •2.7.2. Количественное определение антибиотиков микробиологическим методом
- •2.7.3. Количественное определение кортикотропина
- •2.7.4. Количественное определение фактора свертывания крови VIII
- •2.7.5. Количественное определение гепарина
- •2.7.6. Количественное определение вакцины дифтерии (адсорбированной)
- •2.7.7. Количественное определение вакцины коклюша
- •2.7.8. Количественное определение вакцины столбняка (адсорбированной)
- •2.7.9. Определение функционального состояния Fc-фрагмента иммуноглобулина
- •2.7.10. Количественное определение фактора свертывания крови человека VII
- •2.7.11. Количественное определение фактора свертывания крови человека IX
- •2.7.12. Количественное определение гепарина в концентратах
- •2.7.13. Количественное определение человеческого анти-d-иммуноглобулина
- •2.7.14. Количественное определение антигенной (иммуногенной) активности вакцины гепатита а
- •2.7.15. Количественное определение вакцины гепатита в (rdna)
- •2.7.16. Количественное определение вакцины коклюша (бесклеточной)
- •2.7.17. Количественное определение антитромбина III человека
- •2.7.18. Количественное определение фактора свертывания крови II
- •2.7.19. Количественное определение фактора свертывания крови х
- •2.7.20. Количественное определение инактивированной вакцины полиомиелита in vivo
- •2.7.22. Количественное определение фактора свертывания крови человека XI
- •2.8. Методы анализа лекарственного растительного сырья и лекарственных средств из него
- •2.8.1. Зола, нерастворимая в хлористоводородной кислоте
- •2.8.4. Коэффициент набухания
- •2.8.5. Определение воды в эфирных маслах
- •2.8.10. Растворимость эфирных масел в спирте
- •2.8.11. Определение 1,8-цинеола в эфирных маслах
- •2.8.12. Определение эфирного масла
- •2.8.13. Остаточное количество пестицидов
- •1. Экстракция
- •2. Очистка
- •3. Количественный анализ
- •Относительные времена удерживания инсектицидов
- •2.8.15. Определение показателя горечи
- •2.8.16. Сухой остаток экстрактов
- •2.8.17. Потеря в массе при высушивании экстракта
- •2.9. Фармацевтико-технологические испытания
- •2.9.1. Распадаемость таблеток и капсул
- •2.9.2. Распадаемость суппозиториев и пессариев
- •2.9.3. Тест «растворение» для твердых дозированных форм
- •2.9.4. Тест «растворение» для трансдермальных пластырей
- •2.9.5. Однородность массы для единицы дозированного лекарственного средства
- •2.9.6. Однородность содержания действующего вещества в
- •2.9.7. Прочность таблеток без оболочки на истирание
- •2.9.8. Прочность таблеток на сжатие
- •2.9.9. Измерение консистенции методом пенетрометрии
- •2.9.10 Содержание этанола
- •2.9.11. Испытание на содержание метанола и 2-пропанола
- •2.9.12. Ситовой анализ
- •2.9.15. Насыпной объем
- •2.9.16. Сыпучесть
- •2.9.17. Определение извлекаемого объема парентеральных лекарственных средств
- •Масса действующего вещества высвобожденного при опорожнении
- •Фракция действующего вещества (%)
- •2.9.19. Загрязнение механическими включениями: невидимые частицы.
- •2.9.20. Загрязнение механическими включениями: видимые частицы
- •2.9.21. Загрязнение механическими включениями: метод микроскопии
- •2.9.22. Опредление времени деформации липофильных суппозиториев
- •2.9.23. Определение плотности твердых частиц при помощи пикнометра
- •2.9.24. Устойчивость суппозиториев и пессариев к разрушению
- •2.9.26. Опредедение удельной площади поверхности методом газовой адсорбции
- •III.1.3. Количество образца
- •III.2.1. Метод 1: метод динамического потока
- •III.2.2. Метод 2: метод объёмного анализа
- •2.9.27. Однородность массы одной дозы высвобожденной из многодозового контейнера
- •2.9.28. Определение массы или объема содержимого контейнера для жидких и мягких лекарственных средств
- •3.1. Материалы, используемые для производства контейнеров
- •3.1.1. Материалы, используемые для производства контейнеров для человеческой крови и компонентов
- •3.1.1.1. Материалы на основе пластифицированного поливинилхлорида, используемые для производства
- •3.1.1.2. Материалы на основе пластифицированного поливинилхлорида для трубок, используемых в комплектах для переливания крови и компонентов крови
- •3.1.3. Полиолефины
- •3.1.4. Полиэтилен без добавок для контейнеров для парентеральных и офтальмологических лекарственных средств
- •3.1.5. Полиэтилен с добавками для контейнеров для
- •3.1.6. Полипропилен для контейнеров и укупорочных материалов для парентеральных и офтальмологических лекарственных средств
- •3.1.7. Полиэтиленвинилацетат для контейнеров и трубок для лекарственных средств для парентерального питания
- •3.1.8. Силиконовое масло, используемое в качестве смазывающей добавки
- •3.1.9. Силиконовые эластомеры для укупорочных
- •3.1.10. Материалы на основе непластифицированного поливинилхлорида для контейнеров для неинъекционных водных растворов
- •3.1.11. Материалы на основе непластифицированного поливинилхлорида для контейнеров для твердых лекарственных форм для перорального применения
- •3.1.13. Добавки к пластмассе
- •3.1.14. Материалы на основе пластифицированного поливинилхлорида для контейнеров для водных растворов для внутривенного применения
- •3.1.15. Полиэтилентерефталат для контейнеров для лекарственных средств для непарентерального применения
- •3.2. Контейнеры
- •3.2.1. Стеклянные контейнеры для фармацевтического использования
- •3.2.2. Пластмассовые контейнеры и укупорочные средства для фармацевтического использования
- •3.2.2.1. Пластмассовые контейнеры для водных растворов для парентерального применения
- •3.2.3. Стерильные пластмассовые контейнеры для человеческой крови и ее компонентов
- •3.2.4. Пустые стерильные контейнеры из пластифицированного поливинилхлорида для человеческой крови и ее компонентов
- •3.2.5. Стерильные контейнеры из пластифицированного поливинилхлорида для человеческой крови, содержащие раствор антикоагулянта
- •3.2.6. Комплекты для переливания крови и компонентов крови
- •3.2.8. Стерильные одноразовые пластмассовые шприцы
- •3.2.9. Резиновые укупорочные средства для контейнеров, предназначенных для водных лекарственных средств для парентерального применения, порошков и лиофилизированных порошков
- •4. Реактивы
- •4.1. Реактивы, эталонные растворы, буферные растворы
- •4.1.1. Реактивы
- •4.1.2. Эталонные растворы для испытаний на предельное содержание примесей
- •0,1 М фосфатный буферный раствор рН 8,0. 4008400.
- •4.2. Реактивы, титрованные растворы для объемного нализа
- •1 М щелочной раствор меди-этилендиамина. 3008700
- •5.1 Общие тексты по стерилизации
- •5.1.1. Методы приготовления стерильных продуктов
- •5.1.2. Биологические индикаторы стерилизации
- •5.1.3. Эффективность антимикробных консервантов
- •24 Часа
- •5.1.4. Микробиологическая чистота лекарственных средств
- •5.1.5 .Применение f0 концепции при стерилизации паром водных растворов.
- •5.2. Общая информация о вакцинах
- •5.2.1. Общепринятая терминология
- •5.2.2. Стаи кур, не имеющих конкретных патогенов и используемые для производства вакцин и контроля их качества
- •5.2.3. Субстраты клеток для производства вакцин, используемых людьми
- •5.2.6. Оценка безопасности вакцин
- •5.2.7. Оценка эффективности вакцин
- •5.2.8. Снижение риска передачи возбудителей губчатой энцефалопатии через лекарственные средства
- •1. Общие замечания
- •2. Область применения общей главы
- •3.1. Животные как источник материала
- •3.2. Части тел животных, жидкости и выделения в качестве исходных материалов
- •3.3. Проверка процесса
- •5.3. Статистические методы обработки результатов анализа
- •5.3.1. Статистический анализ результатов биологических исследований и количественных определений
- •1.1. Общие положения и точность
- •2. Рандомизация и независимость конкретных исследований
- •3. Количественные определения, основанные на количественных эффектах
- •3.1. Статистические модели
- •3.2. Модель параллельных линий
- •3.2.2.1 Схема полной рандомизации
- •3.2.2.2 Схема рандомизированных блоков
- •3.3. Модель угловых коэффициентов
- •3.3.5.2 (/7С/)-схема
- •4. Тесты с альтернативным типом эффекта 4.1. Введение
- •4.2. Метод пробит-анализа
- •5.1. Модель параллельных линий.
- •5.2. Модель угловых коэффициентов
- •5.3. Альтернативные эффекты
- •6 Объединение результатов количественного определения 6.1. Введение
- •6.2. Взвешенное объединение результатов количественного определения
- •6.3. Невзвешенное объединение результатов количественного опре- деления
- •6.4. Пример определения взешенной средней активности с доверительн1м интервалом
- •7. Дополнение
- •7.1. Общие линейные модели
- •7.4. Ошибки корреляции
- •8. Таблицы и процедуры генерирования
- •8.5. Случайные размещения
- •8.6. Латинские квадраты
- •9. Принятые обозначения
- •1. Выборка
- •1.1. Среднее зна чение и дисперсия
- •1.3. Доверительные интервалы и оценка их величины.
- •1.4. Односторонние и двусторонние доверительные интервалы.
- •2. Метрологические характеристики методики анализа
- •2.1.1. Объединенная дисперсия и объединенное среднее
- •2.1.2. Критерий Бартлетта.
- •2.1.3. Критерий Кохрейна.
- •2.2. Проверка наличия значимой систематической погрешности.
- •3. Сравнение двух методик анализа по воспроизводимости
- •4. Метрологическая характеристика среднего результата.
- •5. Сравнение средних результатов двух выборок
- •5.3. Известно точное значение величины а.
- •6. Интерпретация результатов анализа, полученных с помощью метрологически аттестованной методики.
- •6.1. Оценка сходимости результатов параллельных определений.
- •6.2. Определение необходимого числа параллельных определений.
- •6.3. Гарантия качества продукции.
- •7. Расчет и статистическая оценка параметров линейной зависимости
- •8. Последовательная схема статистического анализа результатов химических измерений
- •9. Примеры
- •9.1 Вычисление среднего значения и дисперсии.
- •9.2 Проверка однородности выборки малого объема
- •9.3. Вычисление доверительных интервалов и неопределенностей измерений.
- •9.4. Проверка гипотезы равенства дисперсий.
- •9.4.1. Объединение результатов выборок разного объема.
- •9.4.2. Объединение результатов выборок одинакового объема.
- •9.5. Сравнение двух методик анализа по воспроизводимости.
- •9.6. Сравнение средних результатов двух выборок.
- •9.7. Оценка качества продукции.
- •9.8. Контроль содержания салициловой кислоты в салициловом спирте посредством секвенционального анализа.
- •10. Расчет неопределенности функции нескольких случайных переменных
- •10.1. Линейная модель
- •10.1.1. Взвешенное среднее
- •10.2. Подход Уэлча-Сатертуэйта
- •10.3. Примеры расчетов неопределенности функции нескольких переменных
- •10.3.1. Расчет неопределенности вэжх-анализа готового лекарственного средства
- •10.3.1.1. Конечная аналитическая операция
- •10.3.1.2. Суммарная неопределенность пробоподготовки asp,r.
- •10.3.1.3. Расчет суммарной неопределенности анализа aAs,r
- •10.3.2. Прогноз неопределенности спектрофотометрического анализа готового лекарственного средства
- •10.3.3. Расчет среднего значения нескольких неравноточных выборок
- •1. Введение
- •2. Аналитические испытания и методики, подлежащие валидации
- •3. Валидационные характеристики и требования
- •4. Словарь
- •2. Специфичность
- •5. Правильность
- •5.1. Количественное определение
- •5.2. Примеси (количественное содержание).
- •7. Предел обнаружения
- •8. Предел количественного определения
- •8.3. Использование калибровочной прямой и стандартного отклонения сигнала
- •9. Робастность
- •10. Проверка пригодности хроматографической системы
- •3. Неинструментальные испытания на чистоту и предельное содержание примесей
- •5. Разделительные методы
- •6.1. Метод добавок
- •6.2. Сравнение с арбитражным методом
- •5.4. Остаточные количества органических растворителей
- •5.4.1. Введение
- •5.4.2. Область применения
- •5.4.3. Общие положения
- •5.4.4. Предельные содержания остаточных растворителей
- •5.5. Алкоголеметрические таблицы
- •5.6. Отчет об исследовании интерферонов
- •3.3. Процедура исследования
- •3.3.1. Определение уровня доза-ответ
- •5.7. Таблица физических упоминаемых в фармакопеи
- •Вероятность эмиссии
- •Энергия (мЭв)
- •Энергия (мЭв)
- •Вероят ность эмиссии (на
- •Энергия (мЭв)
- •Вероятность эмиссии
- •5.8. Биодоступность и биоэквивалентность генерических лекарственных средств
- •3. Регистрационная оценка взаимозаменяемых лекарственных
- •4. Исследования эквивалентности, необходимые для
- •4.2.1. Исследования биоэквивалентности/биодоступности (исследования на человеке)
- •4.2.2. Общие методические подходы к выполнению исследований биоэк- вивалентности/биодоступности
- •4.2.3. Исследования сравнительной кинетики растворения (исследования вне живого организма)
- •4.3. Отсутствие необходимости в исследованиях биоэквивалентности или биодоступности
- •5. Дизайн и проведение исследований биологической эквивалентности и биодоступности на людях 5.1. Общие требования.
- •5.2. Испытуемые
- •6. Регламент фармакокинетического исследования
- •7. Аналитический метод
- •8. Анализ фармакокинетических данных
- •8.1. Параметры, подлежащие оценке
- •8.1.1. Однократное введение лекарственного средства
- •8.1.2. Многократное введение лекарственного средства
- •9. Исключение резко выделяющихся наблюдений
- •12. Фармакодинамические исследования
- •13. Клинические испытания
- •14. Тест сравнительной кинетики растворения in vitro
- •15. Клинически значимые колебания биодоступности, обуславливающие отказ в регистрации лекарственного средства
- •Лабораторных животных
- •Участие в испытаниях биоэквивалентности/биодоступности
- •Номограмма для определения достаточного числа добровольцев по результатам проведенного исследования.
- •Хорошо растворимые лекарственные средства
- •Средства с высокой степенью абсорбции
- •Перечень терапевтических (лечебных) доз средств на основе лекарственного растительного сырья
- •Основная литература
- •6. Общие статьи на лекарственные формы и субстанции
6. Регламент фармакокинетического исследования
Общая схема исследований
Исследования лекарственных средств осуществляется по слепой рандомизированной перекрестной сбалансированной схеме, включающей два периода и две последовательности - каждый испытуемый последовательно получает исследуемый препарат (Т) и препарат сравнения (R) или наоборот (схема «RT/TR»).
Однако, схема исследования зависит от типа лекарственного средства и в ряде случаев более пригодными могут быть иные схемы. Такое изменение определяется исследователем по согласованию со спонсором, вносится в программу (протокол) исследования и утверждается Министерством здравоохранения.
Число испытуемых
В исследование должно быть включены испытуемые, в количестве достаточном для обеспечения статистической значимости исследования. При этом мощность статистического теста дня проверки биоэквивалентности должна поддерживаться на уровне не меньше 80% для выявления 20%-ных различий между показателями сравнения.
Минимальное число включенных испытуемых 18 человек. Большее число испытуемых может потребоваться для сравнения препаратов, обладающих значительной вариабельностью фармакокинетических параметров.
При планировании эксперимента априорные значения вариации основных показателей сравнения (AUC и Стах), необходимые для расчета числа испытуемых, могут быть оценены по результатам сходных исследований, литературным данным, результатам пилотного исследования, а предварительная оценка необходимого числа испытуемых может быть получена с помощью таблицы 1 или номограммы приложения 4.
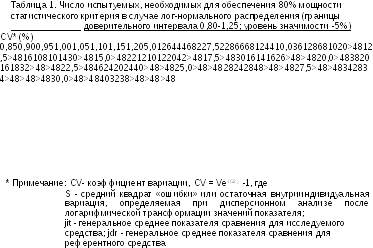 Если
при проведении статистического сравнения
уровень мощности теста оказался ниже
80%, в тех случаях, когда сравниваемые
препараты оказались небно-эквивалентными,
для принятия обоснованного заключения
о небиоэквивалентности необходимо
включить в исследование большее число
испытуемых.
Если
при проведении статистического сравнения
уровень мощности теста оказался ниже
80%, в тех случаях, когда сравниваемые
препараты оказались небно-эквивалентными,
для принятия обоснованного заключения
о небиоэквивалентности необходимо
включить в исследование большее число
испытуемых.
Интервал времени между приемом лекарственных средств
Интервал времени между приемом препаратов зависит от длительности циркуляции лекарственного средства в организме, определяемой периодом полуэлиминации (ТД и должен составлять не менее 6Ту2.
Достаточность отмывочного периода может быть определена исходя из определения остаточных концентраций лекарства в нулевой точке перед началом второго этапа исследования, которые должны составлять не более чем 5% от величины Стах первого этапа исследования.
Схема отбора проб
Схема отбора проб определяется формой кривой «концентрация действующего вещества - время». Выбор моментов временя отбора проб должен обеспечивать получение нескольких точек для каждого фрагмента фармакокинетической кривой -не менее 2 для фазы первоначального возрастания концентрации, 2 точки вблизи планируемого времени достижения Стах и не менее 3-4 точек для фазы элиминации.
Общая продолжительность наблюдения за концентрацией действующего вещества при однократном приеме лекарственного средства должна быть не менее чем в 4 раза больше периода полувыведения, а при многократном приеме препарата - равной интервалу дозирования. При однократном приеме длительность наблюдения считается удовлетворительной, если для усредненного фармакокинетического профиля величина площади под кривой «концентрация - время» в пределах от нуля до момента отбора последней пробы составляет не менее 80% от полной площади.
Исследование метаболитов
Использование данных о продуктах обмена в исследовании биологической эквивалентности требует осторожного подхода. Оценка биологической эквивалентности основывается в основном на измерении концентраций фармакологически активного вещества и его активных метаболитов, если таковые имеются.
Измерение концентрации метаболита исходного фармацевтического ингриди-ента может потребоваться в следующих случаях:
если исследуемое лекарственное средство содержит исходный фармацевтический ингредиент в виде пролекарства;
если метаболит вносит весомый вклад (не менее 50%) в профиль безопасности или клинической эффективности данного лекарственного средства;
если невозможно измерить концентрацию активного лекарственного вещества ввиду слишком низких его концентраций в крови, плазме или сыворотке в течение требуемого времени с точки зрения возможности достоверного аналитического определения, или если исходный фармацевтический ингредиент является нестабильным в биологических жидкостях.
Если производится измерение в моче, определяемый метаболит должен предоставлять собой основную фракцию дозы. Если измерения концентрации активных метаболитов обычно считается допустимым, то определение концентрации неактивного метаболита лишь в редких случаях можно считать обоснованным.