


Доступ к рецептору для μ-агонистов зависит от следующих физикохимических и общих фармакологических характеристик:
1)липофильность;
2)величина рКа;
3)степень ионизации;
4)доля несвязанного препарата;
5)явный объем распределения;
6)клиренс;
7)пути введения препарата.
Таким образом, различия в клиническом действии отдельных опиоидов на самом деле отражают особенности физико-химических и фармакокинетических характеристик разных препаратов, а не различия их фармакодинамики. Эта концепция крайне важная для правильного клинического использования опиоидов. Так, все опиоиды способны обеспечить одинаковый обезболивающий эффект, быть одинаково действенными при соответствующей
коррекции их дозировки с учетом путей введения и физико-химических и фармакологических характеристик (рис. 8-7, табл. 8-8). Эта концепция известна как концепция равнообезболиваюгцего дозирования [52, 53].
Схему равнообезболивающего дозирования, изображенную на рис. 8-7 и в табл. 8-8, следует использовать лишь в качестве ориентира. При создании
подобных конверсионных таблиц приходится сталкиваться со множеством методологических проблем. Поэтому практический врач не должен быть догматиком при интерпретации приведенных в ней данных.
Рис. 8-7. Пути назначения препаратов в концепции равнообезболивающей конверсии.
Приведенное конверсионное соотношение является приблизительным, поэтому практикующий врач не должен быть догматиком в интерпретации полученных результатов.
Таблица 8-8. Аналгезические эквиваленты опиоидов
Опиоиды |
Пути введения |
Эквианалгезические до- |
|
|
зы2 |
Морфин |
Парентерально 3 |
10 мг |
|
Энтерально |
30 мг |
Кодеин |
Парентерально 3 |
130 мг |
|
Энтерально |
200 мг |
Оксикодон |
Парентерально 3 |
15 |
мг |
|
Энтерально |
30 |
мг |
Леворфанол |
Парентерально 3 |
2 мг |
|
|
Энтерально |
4 мг |
|
Гидроморфон |
Парентерально 3 |
1,5 мг |
|
|
Энтерально |
7,5 мг |
|
Меперидин |
Парентерально 3 |
75 |
мг |
|
Энтерально |
300 мг |
|
Метадон |
Парентерально 3 |
10 |
мг |
|
Энтерально |
20 |
мг |
Фентанил |
Парентерально 3 |
100 |
мкг |
|
Энтерально |
Не выпускается |
|
1Каждый препарат вводили в однократной дозе внутримышечно, а его действие сравнивали с действием морфина для определения относительной силы. 2Конверсию доз различных опиоидов проводили без учета возможной перекрестной переносимости (неполной устойчивости).
3Соотношение действенности морфина при энтеральном его введении к -па рентеральному в однократной дозировке по поводу острых или послеоперационных болей равно 1 :6. Это соотношение может снизиться до1:3 или 1:2 при повторных введениях препарата (хронические боли). (По Foley, с разре-
шения [52].)
Замысел исследования. Схема равнообезболивающего дозирования традиционно основана на однократном внутримышечном введении опиоида. При этом сравнивают полученный эффект с действием морфина и вычисляют соотношение действенности препаратов [52]. Однако разные режимы введения препаратов обусловливают существенные вариации в величинах - соз дающихся пиков концентрации препаратов и во времени их наступления. (Полное обсуждение ситуаций, встречающихся при рассасывании препаратов из мышечных депо, приведено в гл. 10.) Более практичным является сравнение действенности препаратов при их внутривенном введении.
В клинической практике продолжительную аналгезию редко удается обеспечить однократным введением опиоидов. Более реальным конверсионным ориентиром могла бы стать методика с использованием многократных дозировок. С этой целью проводятся исследования по внутривенной аналгезии, контролируемой самим пациентом (ВВ-АКП). Использование ВВ-АКП устраняет стремление экспериментировать, так как предоставляет пациенту самому определять, когда и сколько опиоидов он должен получить. ВВ-АКП также позволяет определить среднее потребление анальгетиков, несмотря на значительные различия в необходимости обезболивания [55-59].
Неполная перекрестная устойчивость. Развитие устойчивости к обезболивающему действию одного из опиоидов служит основанием для замены препарата. Более выраженный аналгезирующий эффект нового препа-
рата в этих условиях объясняется неполным характером перекрестной устойчивости [60, 61]. Новый опиоид рекомендуют назначать в начальной дозе, равной половине равнообезболивающей. Подобная доза устанавливается эмпирически. Предполагается, что относительная эффективность опиоидов может нарастать при их повторном введении[53]. Необходимо отметить, что в табл. 8-8 не была учтена неполная перекрестная устойчивость.
Соотношение активности при оральном и парентеральном назна-
чении. Биологическое использование морфина при его энтеральном введении варьирует от 15 до 64%, в среднем составляя 38% [62]. Большинство авторов полагают, что при парентеральном введении биологическое действие оказывают все 100% введенной дозы препарата. Поэтому при переходе с эн-
терального на парентеральный путь введения морфина важно правильно учесть особенности первоначального метаболизма этого препарата [63].
В случаях острой боли и при однократном применении отношение активности при оральном назначении морфина к его активности при парентеральном введении равно1:6 [64]. При хронических болях и повторных назначениях морфина это отношение становится 1:2 или 1:3 [62, 65, 66]. Приведенные данные были получены эмпирически и адаптированы в отношении других опиоидов.
Эквивалентность активности при разных путях парентерального введения. Многие конверсионные схемы одинакового обезболивания основаны на предположении, что при подкожном (ПК), внутримышечном (ВМ) и внутривенном (ВВ) введении опиоиды действуют одинаково. Подобное предположение заранее допускает одинаковую биологическую активность независимо от пути введения препарата. Между тем установлены отдельные факты, позволившие многим авторам поставить под сомнение данное допущение.
В исследованиях Urquhart и сотр. [67] установлено, что при подкожном введении гидроморфона (ПК-АКП) для аналгезии требовалось значительно большее количество препарата, чем при внутривенном его введении(ВВАКП). Это наблюдение подтверждает неодинаковую биологическую активность препарата при разных путях его введения. По мнению автора, соотношение активности гидроморфона при ПК и ВВ введении варьирует от1 : 1,5
до 1:2.
Дыхание
Все μ-агонисты и частичный агонист бупренорфин угнетают реакцию дыхательного центра в стволе мозга на повышенную концентрацию двуокиси углерода в крови(РСО2). Степень этого угнетения пропорциональна дозе опиоида. Препараты со свойствами агонист-антагонистов не вызывают столь выраженного угнетающего действия, зависящего от дозы, а оказывают лишь ограниченное влияние (так называемый потолочный эффект) [32]. Опиоиды со свойствами агонистов подавляют дыхательные центры также в области

моста и продолговатого мозга, где регулируется ритм дыхания. Это проявляется удлинением пауз между вдохами, замедленным вдохом и появлением периодического дыхания [68, 69].
Снижение возбудимости дыхательных центров к СО, вызванное μ-
2
агонистами, сопровождается |
увеличением |
показателя РСО2 и смещением |
кривой реакции вправо(рис. |
8-8). Под |
влиянием эквивалентных доз - μ |
агонисты вызывают одинаковые по степени нарушения дыхания и соответствующее смещение кривой реакции на 2СО[70-72]. Агонист-антагонисты
также смещают кривую реакции на СО вправо, но при нарастающих дозах
2
эта кривая приобретает закругленность («форма колокола»).
Рис. 8-8. Кривые реакции дыхания (СО2), полученные до и через1 ч после внутримышечного введения 10 мг морфина.
Эквивалентные обезболивающие дозы опиоидов вызывают сопоставимые изменения реактивности дыхательных центров и одинаковое смещение кривой реакции на СО2 вправо.
Угнетение дыхания, вызванное опиоидами, в клинике выражается снижанием числа дыхательных движений и часто сопровождается увеличением дыхательного объема. Однако подобная компенсация оказывается явно не-
достаточной, что подтверждается ростом РСО. После введения - те
2
рапевтических доз препарата минутный объем дыхания может оставаться сниженным в течение 4-5 ч. Высокие дозы μ-агонистов или частичных агонистов вызывают апноэ. Особенно значительное угнетение дыхания под влиянием опиоидов отмечают у пожилых пациентов во время сна. В то же время боль в послеоперационном периоде противодействует угнетению дыхания опиоидами [71].
Снижение уровня сознания

Опиоиды относятся к препаратам, изменяющим душевное состояние. Больные, которые должны были бы испытывать боли, испытывают ощуще-
ние тепла, говорят о своем хорошем самочувствии, у них появляются сонли-
вость и эйфория. Нарушения душевного состояния и восприятия окружаю-
щего, вероятно, опосредованы лимбической системой [73].
Таблица 8-9. Гипнотическое действие опиоидов (в порядке снижения активности)
Диацстилморфин == гидроморфон Меперидин = налбуфин = пентазоцин Морфин Метадон Кодеин Фентанил
По данным Freye [75].
Иногда опиоиды вызывают сон, но потери сознания не происходит даже при высоких дозах. Повреждающие стимулы могут разбудить больных даже после введения больших(«анестетических») доз препарата [74]. Способность опиоидов вызывать сон значительно варьирует даже при введении эквивалентных обезболивающих доз разных препаратов (табл. 8-9) [75].
Изменения на электроэнцефалограмме под влиянием опиоидов напоминают сдвиги, возникающие во сне. Они заключаются в смене быстрых -а волн более медленными дельта-волнами (рис. 8-9) [76].
Противокашлевое действие
Все опиоиды подавляют кашлевой рефлекс(некоторые частично), оказывая непосредственное влияние на кашлевой центр в продолговатом мозге. Зависимости между угнетением дыхания и подавлением кашля не обнаружено [72]. Способность разных опиоидов подавлять кашлевой рефлекс различна
(табл. 8-10).
Таблица 8-10. Противокашлевой эффект опиоидов (в порядке снижения активности)
Диацстилморфин = фентанил = гидроморфон = гидрокодон Метадон Кодеин Морфин Леворфанол
Меперидин == пентазоцин
По данным Freye [75].

Зрачковый эффект
Большинство μ- и капа-агонистов вызывают сужение зрачка у человека. Миоз служит проявлением возбуждающего действия опиоидов на автоно-м ный сегмент ядра глазодвигательного нерва(сегмент Эдингера-Вест-фаля). После применения высоких доз опиоидов зрачки становятся размером с б- у лавочную головку. Миоз можно устранить атропином.
Тошнота и рвота
Опиоиды вызывают тошноту и рвоту, стимулируя хеморецепторы триггерной зоны продолговатого мозга(area postrema) [72]. Этот эффект от-
ражает действие опиоидов как агонистов допамина и его рецепторов. Кроме того, опиоиды могут провоцировать тошноту и рвоту, замедляя пассаж содержимого по желудочно-кишечному тракту. При назначении в эквивалент-
ных дозах все опиоиды в одинаковой степени вызывают тошноту и рвоту.
Рис. 8-9. Типичный пример сжатого спектрально-рядового анализа одного производного ЭЭГ (TIII-CVIII) во время анестезии суфентанилом15 мкг/кг. Отмечен сдвиг высокой энергии активности. (Из Bovill et al., с разрешения.)

Толерантность, физическая зависимость и наркомания
Продолжительный прием опиоидов в больших дозах приводит к ослаблению их действия. Поэтому требуются все возрастающие количества препа- рата для достижения одной и той же степени аналгезии. Данный феномен, называемый толерантностью, характерен для всех препаратов из группы опиоидов. Толерантность может возникать при назначении любого из них и сопровождаться перекрестной устойчивостью к действию также и остальных опиоидов. Часто подобная толерантность бывает неполной[7]. Толерант-
ность совсем не обязательно сопровождается физической зависимостью. Физическая зависимость не является синонимом наркомании. Это фи-
зиологическое состояние, проявляющееся синдромом отмены после прекра- щения приема опиоидов (абстиненция). Начальный период синдрома отмены сопровождается сонливостью, зевотой, потливостью, насморком, тахикардией. Схваткообразные боли в животе, тошнота и рвота достигают максимальной силы через 72 ч после отмены препарата. Во время проявлений синдрома отмены толерантности к опиоидам нет.
Всемирной организацией здравоохранения (ВОЗ) дано следующее определение наркомании: «Психическое, а иногда и физическое состояние в ре- зультате взаимодействия человеческого организма и препарата, характеризующееся поведенческими и другими реакциями, всегда включающими внутреннее принуждение к непрерывному или периодическому приему пр-е парата с целью испытать его действие на психику, иногда для устранения дискомфорта, вызванного отсутствием препарата». Толерантность при этом может наблюдаться или нет [77].
Это определение тесно примыкает к распространенному представлению о наркомании как о принуждении или очень сильном стремлении испытать действие препарата на психику. Наркомания включает компульсивное поведение и психологическую зависимость. Она отличается от толерантности по концептуальным и феноменологическим характеристикам(фармакологические свойства препаратов этой группы), а также от физической зависимости (физиологический эффект свойствен препаратам этой группы).
Обычно спрашивают, может ли назначение опиоидов с лечебной целью (например, послеоперационная аналгезия) индуцировать наркоманию? Частота развития наркомании была изучена у12000 госпитальных больных, получавших не менее одного из сильнодействующих опиоидов. Только у 4 из них в последующем развилась наркомания. Следовательно, морфинизм редко бывает следствием медицинских мероприятий [78].
Тенденция опиоидов вызывать наркоманию в определенной степени связана с их обезболивающим действием(табл. 8-11). Только агонист- антагонистические опиоиды значительно менее опасны в этом отношен,ии возможно, именно из-за антагонистического компонента их смешанного действия [75]. Привыкание к опиоидам определяется не только психогенным действием препаратов. Продолжительный прием опиоидов без развития нар-
комании продемонстрирован у пациентов с болями на почве неопухолевого поражения. Прием препаратов сам по себе еще не является главным факт- о ром в развитии наркомании. Более важными моментами в ее возникновении могут быть предшествующие личностные особенности, социальное окруже-
ние и финансовые возможности [81].
Таблица S-11. Потенциал наркотической зависимости опиоидов(в порядке снижения потенциала)
Диацетилморфин = оксиморфон фентанил = метадон Лtворфанол= морфин Меперидин Кодеин
Налбуфин =пентазоцин
По данным Freye [75].
Действие на желудочно-кишечный тракт
Желудок, тонкий и толстый кишечник
Опиоиды подавляют продольную перистальтику тонкого и толстого кишечника, замедляя пассаж его содержимого. Одновременно усиливается поперечная перистальтика, проявляющаяся как ритмичные сегментарные сокращения разных отделов кишечника. Двигательная активность желудка снижается, а тонус его антрального отдела усиливается. Замедленному опорожнению желудка способствует также повышение тонуса начального отдела двенадцатиперстной кишки. Под влиянием опиоидов нарастает тонус привратника, анального сфинктера и илеоцекального клапана. Все эти изменения, сочетающиеся с ослаблением продольной перистальтики, значительно удлиняют время пассажа содержимого по кишечнику. Длительное прохождение содержимого в свою очередь способствует усиленному всасыванию воды из кишечника, повышению вязкости и плотности каловых масс. Все это приводит к развитию запоров.
Механизм действия. Тонкости механизмов влияния опиоидов на перистальтику кишечника остаются неясными. В них сочетаются факторы локального и центрального воздействия. Опиоиды влияют на холинергические, серотонинергические и энкефалинергические рецепторы нервных сплетений в мышечном слое стенки кишечника. Предотвратить действие опиоидов на кишечник в условиях эксперимента на животных не удается ни назначением ганглиоблокирующих агентов, ни исключением внешней иннервации. Введение опиоидов в область спинного мозга в такой же степени подавляет -пе ристальтику (см. гл. 11 и 26). Подвижность кишечника ослабевает даже при

введении препарата в желудочки мозга. Правда, внутривенное введение налоксона или ваготомия устраняют этот эффект[82]. Таким образом, приведенные сведения убеждают, что снижение кишечной моторики, связанное с приемом опиоидов, опосредовано как локальными, так и центральными механизмами.
Желчные пути
Под влиянием μ-агонистов давление в желчных путях резко возрастает. После подкожного введения 10 мг морфина давление в общем желчном протоке увеличивается в 10 раз [72]. Опиоиды из группы агонист-антагонистов воздействуют на давление в желчных путях гораздо слабее, чем μ-агонисты [84]. Налбуфин снимает спазм сфинктера Одди и устраняет болевые ощущения (аналгезия, опосредованная капа-рецепторами) [85].
Действие на сердечно-сосудистую систему
Опиоиды используют как средства анестезии во время операций, рас- сматривая их как препараты, не ухудшающие функцию сердечно-сосудистой системы. Между тем эти препараты оказывают хронотропное, инотропное действие и влияют на периферические сосуды. Опиоиды в зависимости от дозировки вызывают более или менее выраженную брадикардию вследствие стимуляции ядра блуждающего нерва в продолговатом мозге. Это действие может быть блокировано атропином[86-88]. Меперидин благодаря своему структурному сходству с атропином вызывает тахикардию (рис. 8-10).
Опиоиды, за исключением меперидина, в клинических дозах не подавляют сократительную способность миокарда. Отрицательное инотропное действие меперидина проявляется при его введении в дозах2-2,5 мг/кг. Все опиоиды способны оказывать угнетающее влияние на миокард, но этот эффект в условиях клинической анестезиологии не достигается даже при назначении высоких доз [89-92].
Морфин оказывает как непосредственное, так и опосредованное влияние на гладкие мышцы сосудов. Опосредованное влияние связано со стимуляцией выделения гистамина, расширяющего артерии и вены. Именно этот медиатор играет ведущую роль в расширении сосудов. Меперидин и кодеин также высвобождают гистамин, в то время как фентанил и суфентанил не оказывают подобного действия [93-96].

Рис. 8-10. Меперидин в высоких дозах может вызывать тахикардию вследствие его структурного сходства с атропином.
μ-АГОНИСТЫ Алкалоиды Морфин
Морфин служит прототипом агониста (рис. 8-11). Как указано выше, он относится к опиатам, т. е. к алкалоидам, получаемым из самого опиумного мака. Это растение все еще остается основным источником морфина, поскольку его химический синтез затруднен.
Фармакокинетика. После внутривенного введения морфин быстро распределяется в органах и тканях. Уже через 10 мин 96-98% этого препарата исчезает из плазмы крови. Объем распределения морфина относительно велик, что указывает на его интенсивное поглощение тканями, в том числе скелетными мышцами [97, 98].
Stanski и сотр. [98] показали, что пик концентрации препарата в крови наступает через 7,5-20 мин после его внутримышечного введения. Brunk и Delle [99] нашли, что в период от 15 мин до 3 ч концентрация морфина в крови поддерживается на более высоком уровне после внутримышечного и подкожного введения, чем после внутривенной инфузии. Очевидно, в последнем случае морфин быстрее выходит из плазмы, а при внутримышечном и подкожном введении создаются депо препарата, служащие источником его последующего поступления в кровь.
Быстрое исчезновение морфина из плазмы при внутривенном введении приводит к тому, что уровень его в крови не коррелирует с фармакологическим действием [100, 101]. Относительно замедленное проникновение морфина через гематоэнцефалический барьер можно объяснить его гидрофильностью. Пик концентрации морфина в цереброспинальной жидкости наступает через 15-30 мин после его внутривенной инфузии, а последующее снижение уровня происходит медленнее, чем соответствующее падение концентрации препарата в крови. Таким образом, аналгетическое действие морфина может не иметь явной связи с пиком его концентрации в крови после внутривенного введения.

Метаболизм: значение глюкуронидовых метаболитов. Основной путь метаболизма морфина - это его глюкуронизация, происходящая в печени, а также в других органах, например в почках [102]. Основные метаболиты морфина - морфин-3-глюкуронид и морфин-6-глюкуронид (рис. 8-12) [62,
103]. Морфин-6-глюкуронид обладает выраженными аналгезирующими свойствами и, возможно, в определенной степени определяет обезболивающее действие препарата. Этот метаболит в45 раз активнее морфина при внутримозговом введении и в 4 раза - при подкожном введении [104].
Рис. 8-12. Метаболизм морфина.
Основными метаболитами являются морфин-3-глюкуронид и морфин- 6-глюкуронид. Последний обладает выраженным аналгезирующим действием и в значительной степени обусловливает обезболивающее влияние морфина. Оба метаболита выделяются с мочой и могут аккумулироваться при повторном назначении у больных с почечной недостаточностью. Деметилирование играет незначительную роль в метаболизме морфина.
Деметилирование играет незначительную роль в метаболизме морфина. Этому типу метаболизма подвергается около5% введенной дозы препарата, преобразующегося в норморфин [105]. Проявлением обычного метаболизма морфина может быть также продукция небольшого количества кодеина.
Печень служит главным органом, осуществляющим метаболизм морфина [106]. Печеночная недостаточность почти не отражается на процессах глюкуронизации, и морфин хорошо переносится пациентами вплоть до развития состояния прекомы [107, 108].
Метаболиты морфина выводятся в основном с мочой, с желчью выделяется не более 7-10% продуктов его обмена. Менее 10% морфина выделяется почками в неизмененном виде. Процессы выведения не нарушаются даже у больных с почечной недостаточностью [62, 101, 109], однако активный метаболит (морфин-6-глюкуронид) может кумулироваться при снижении выделительной функции почек. Это вызывает продление действия, седацию и угнетение дыхания [110-114]. Поэтому у больных с почечной недостаточностью для обезболивания следует выбирать не опиоиды, а другие препараты.
Фармакологическое действие. Как указано выше, морфин является прототипом опиоидов. Основные его фармакологические свойства уже обсуждены в предыдущих разделах этой главы.
Использование в клинике и фармацевтические препараты. Морфин выпускают в виде гидрохлорида и сульфата. Для энтерального употребления имеются таблетки быстрого и замедленного действия, а также эликсир. Метаболизм принятого энтерально морфина проходит по первому из указанных выше путей, в общую циркуляцию попадает около30% принятой дозы препарата. Несмотря на это, повторное назначение препаратов медленного действия обеспечивает хорошее обезболивание при выраженных и тяжелых болях хронического характера. Препараты быстрого действия используются для устранения внезапно возникающих болей и обеспечивают дополнительную аналгезию [115]. Морфин можно назначать ректально в свечах, а также в виде инъекций.
Кодеин
Кодеин это натуральный алкалоид группы морфина. Он образуется при
субституции метиловой группы в С молекулы морфина(рис. 8-13). В ре-
3
зультате подобного замещения снижается метаболизм этого препарата в печени (метаболизм первого прохождения), что усиливает эффект действия кодеина.
Фармакокинетика. После всасывания кодеин подвергается метаболизму в печени (преимущественно деметилирование с образованием норкодеина) и затем выделяется почками. В отличие от морфина он выводится преимущественно в неактивной форме. Около 10% введенной дозы кодеина после деметилирования трансформируется в морфин. Именно данная фрак-

ция объясняет обезболивающее действие кодеина, поскольку сам он обладает очень слабым аффинитетом к опиоидным рецепторам [73, 116].
Рис. 8-13. Кодеин синтезируется путем подстановки метильной группы к С3 в молекуле морфина. Кодеин относится к алкалоидам, встречающимся в природе. Он значительно активнее при энтеральном приеме, так как в незначительной степени подвержен метаболизму при первичном прохождении.
Фармакологическое действие. Кодеин обладает слабым или умеренным обезболивающим действием, его не следует применять при выраженных болях. Точно так же ограничена его способность вызывать седативный -эф фект, тошноту, рвоту и угнетать дыхание. Кодеин выпускают и для инъекций, но внутривенное введение препарата не рекомендовано, поскольку его способность стимулировать высвобождение гистамина выражена даже сильнее, чем у морфина. Вероятность развития наркотической зависимости при приеме кодеина очень невелика (см. табл. 8-11).
Использование в клинике и фармацевтические препараты. Энте-
ральный прием кодеина в дозе15 мг оказывает выраженное противокашлевое действие. При повышении дозы до 60 мг этот эффект усиливается [117]. Кодеин обычно включают в состав комплексных препаратов, используя его противокашлевое действие, а также часто комбинируют с неопиоидными анальгетиками для снятия легких или умеренных болей [118]. Максимальный обезболивающий эффект отмечают при дозе60 мг, эквивалентной 650 мг ацетилсалициловой кислоты (аспирин). При внутримышечном введении 130 мг кодеина эквивалентны 10 мг морфина.
Полусинтетические опиоиды
Эта группа опиоидов объединяет препараты, получаемые синтетическим путем при простой химической модификации молекулы морфина(см. рис. 8-2). В природных условиях данные препараты не встречаются.
Диацетилморфин (героин, диаморфин)
Диацетилморфин (рис. 8-14) это представитель препаратов, не связывающихся с опиоидными рецепторами и не оказывающих аналгезирующего действия. Он подвержен быстрому гидролизу с образованием6-моно- ацетилморфина и морфина. Фармакологический профиль диацетилморфина очень похож на профиль морфина и не имеет преимуществ перед ним при

внутримышечном или энтеральном введении [119, 120]. До сих пор не установлено, есть ли у диацетилморфина какие-либо преимущества перед морфином при внутривенном, эпидуральном или субарахноидальном при менении. Диацетилморфин запрещен к производству и применению вСША из-за его способности быстро вызывать наркотическую зависимость.
Рис. 8-14. Диацетилморфин относится к продуктам, быстро гидролизирующимся в плазме с образованием моноацетилморфина(обладающего болеутоляющим действием) и морфина.
Гидроморфон (дилаудид)
Гидроморфон (см. рис. 8-2) при парентеральном введении действует примерно в 7-8 раз сильнее морфина. Клинико-морфологический его профиль такой же, как у морфина [121, 122]. Есть не внушающие большого доверия сообщения о меньшей частоте побочных реакций(тошнота, рвота, угнетение дыхания, ретенция мочи и запоры), однако подтверждений подобных сведений мало [121].
Фармакокинетика. Несмотря на многолетнее применение гидроморфона в клинике, фармакокинетика этого препарата изучена недостаточно [123]. Недавно было установлено, что скорость распределения препарата в тканях такая же, как и у морфина. Около 90% препарата исчезают из плазмы крови уже через 10 мин после его введения. Выделение гидроморфона, как и морфина, зависит от его быстрого потребления тканями с последующим медленным выходом обратно в кровь [124].
Использование в клинике и фармацевтические препараты. Гидро-
морфон в отличие от морфина, кодеина и меперидина не подвергается метаболизму с образованием норгидроморфона[116]. Эта особенность делает применение гидроморфона особенно целесообразным у пациентов с почечной недостаточностью. В остальных отношениях фармакологический профиль гидроморфона почти не отличается от такового морфина, что не позволяет говорить о его преимуществах.
Гидроморфон выпускают в таблетках, содержащих от 1 до 4 мг препарата. Растворы для инъекций содержат его в дозах1, 2 и 4 мг/мл. Внутримы-

шечное введение 1,5 мг гидроморфона оказывает действие, эквивалентное 10 мг морфина. Аналгезирующий эффект продолжается 3-5 ч. Обезболивающее действие препарата при энтеральном приеме в5 раз слабее, чем при внутримышечном введении. Имеются свечи с гидроморфоном для ректального вве-
дения [125, 126].
Оксиморфон (нуморфан)
Оксиморфон синтезируют путем присоединения гидроксильной группы к С14 в молекуле гидроморфона (см. рис. 8-2). При парентеральном введении оксиморфон примерно в 10 раз активнее морфина [127, 128]. Соотношение активности оксиморфона при энтеральном и парентеральном назначении составляет 1:6 [128]. Клинико-фармакологический профиль в остальных отношениях такой же, как у морфина. Отмечается быстрое развитие наркотической зависимости (такое же, как у героина) и слабое высвобождение гистамина (см. табл. 8-11) [129, 130]. Отдельные сообщения о большой частоте побочных реакций (тошнота и рвота) большинством исследователей подтверждены не были.
Важное значение имеет структурное сходство между оксиморфоном и налоксоном (рис. 8-15). Налоксон (опиоидный антагонист) представляет собой N-аллил-(—СН2—СН=СН2)-замещенный аналог оксиморфона. Структурное сходство этих двух опиоидов используют для изучения взаимоотношений между структурой и активностью, для уточнения взаимодействия агонистов, антагонистов и рецепторов, а также для синтеза новых агонистов
[131 134].
Рис. 8-15. Налоксон (антагонист опиоидов) является N-аллил-(—СН2— СН=СН2)-замещенным аналогом оксиморфона, мощного μ-агониста.
Фармакокинетика. Фармакокинетика оксиморфона изучена недостаточно Менее 10% введенной дозы препарата выводится мочой в неизмененном виде [135].
Использование в клинике и фармацевтические препараты. Прово-
дились обширные исследования по использованию оксиморфона для -под кожной и внутривенной АКП[136-139]. Высокая активность препарата, кратковременность его действия и возможность энтерального приема сделала
перспективными исследования по трансдермальному введению оксиморфона [140]. Обычная доза при чрескожном и внутримышечном введении оксиморфона равна 1-1,5 мг каждые 4-6 ч. При внутривенной инфузии начальная доза составляет 1,5 мг.
Выпускают оксиморфон в ампулах, содержащих 1 или 1,5 мг/мл вещества. Имеются также ректальные свечи, активность препарата в которых в 10 раз меньше, чем активность той же дозы при внутримышечном введении
[141].
Гидрокодон (гикодан, лортаб, викодин, туссионекс)
Клинико-фармакологический профиль гидрокодона такой же, как у кодеина. Он хорошо всасывается при энтеральном приеме(около 50%) и оказывает выраженное противокашлевое действие(см. табл. 8-10). Гидрокодон используют только энтерально обычно в комбинации с другими неопиоидными анальгетиками для получения обезболивающего эффекта.
Фармакокинетика. Гидрокодон подвергается 0-демегилированию, N- деалкализации и 6-кеторедукции [142]. Полагают, что при метаболизме в печени он может образовать гидроморфон. Последнее обстоятельство способно объяснить два описанных в литературе случая смерти после приема гидрокодона в качестве противокашлевого средства [143].
Использование в клинике и фармацевтические препараты. Гидро-
кодон обычно выпускают в комбинации с ацетаминофеном или с ацетилсалициловой кислотой. Подобное сочетание обеспечивает синергизм действия, уровень обезболивания соответствует действию удвоенной дозы каждого из этих препаратов. Побочные же эффекты при этом ослабевают [144].
Оксикодон (перкоцет, перкодон, роксицет, роксикодон, тилокс)
Фармакологический профиль оксикодона такой же, как у морфина. Подобно кодеину и гидрокодону, оксикодон хорошо всасывается после приема внутрь, оказывая обезболивающее действие в течение не менее получаса. Оксикодон выпускают обычно в комбинации с другими анальгетиками неопиоидного ряда. Оксикодон не используют как противокашлевое средство. Кроме того, препарат обладает значительным потенциалом в отношении наркотической зависимости [145].
Фармакокинетика. Фармакокинетика оксикодона, как и большинства других полусинтетических опиоидов, изучена недостаточно. Продуктом его метаболизма является нороксикодон [146, 147].
Использование в клинике и фармацевтические препараты. В США оксикодон назначают только энтерально. При этом его действие в 4 раза сла-
бее, чем у гидрокодона. Препараты для парентерального применения в США не используют. Продолжается изучение средств для трансдермального вве-
дения [148].
Синтетические опиоиды
Леворфанол (лево-дроморан)
Леворфанол единственный доступный μ-агонист из группы морфина (см. рис. 8-3; рис. 8-16). Правовращающий изомер (декстрометорфан) обладает такой же противокашлевой активностью, как кодеин, но не оказывает обезболивающего действия и не вызывает наркомании.
Фармакологический профиль леворфанола такой же, как у морфина, он подвержен значительному метаболизму при первом прохождении, хотя возможны индивидуальные отклонения [149, 150]. Соотношение активности при оральном и парентеральном назначении такое же, как у кодеина и оксикодона. Леворфанол в 7 раз активнее морфина при энтеральном и в5 раз при парентеральном введении (см. табл. 8-8).
Фармакокинетика. Леворфанол быстро рассасывается после подкожного введения. Максимальная аналгезия наступает через60-90 мин, а продолжительность обезболивания такая же, как после парентерального введения морфина. Метаболизм препарата осуществляется медленно (время полувыведепия 11 ч). Поэтому повторные инъекции препарата через короткие интервалы времени могут вызвать его аккумуляцию [150].
Рис. 8-17. Меперидин.
Рис. 8-16. Леворфанол. Обезболивающими свойствами обладает только его левовращающий изомер.
Использование в клинике и фармацевтические препараты. Лево-
рфанол чаще всего используют при хронических болях, преимущественно при ведении больных раком [52, 151].
Выпускают препарат в виде леворфанола гартарата, в таблетках по 2 мг или в растворе для инъекций с концентрацией вещества 2 мг/мл.
Меперидин (демерол)
Меперидин - один из препаратов группы фенилпиперидина(рис. 8-17) относится к опиоидным μ-агонистам. Преимущественно используемые опиоидные анестетики фентанил, суфенганил и алфентанил (рис. 8-18, 8-19 и 8- 20) являются аналогами меперидина.
Фармакокинетика. При энтеральном назначении препарат подвергается метаболизму в печени, и биологическое действие оказывает 45-75% введенной дозы. Меперидин всасывается медленнее, пик его концентрации в крови наступает через 2 ч после приема внутрь [152].
Скорость рассасывания препарата при внутримышечном введении весьма вариабельна, поэтому обезболивающее его действие неустойчивое и часто недостаточное [54, 153].
После внутривенного введения меперидин переходит из крови в ткани, распределение его завершается через 30-45 мин, что гораздо медленнее, чем у морфина (10 мин после внутривенного введения).
Время полувыведения меперидина составляет 3-4,4 ч [154]. Около 60% препарата связывается белками плазмы. Ослабление связывания препарата белками крови у пожилых лиц может привести к увеличению содержания свободной фракции меперидина и вызвать повышенную чувствительность к нему [155].

Рис. 8-21. Метаболизм нормеперидина в печени.
Метаболизм: значение метаболита нормеперидина. Меперидин ин-
тенсивно разрушается в печени(рис. 8-21). Примерно 90% всей введенной дозы подвергается N-деметилированию с образованием нормеперидина и гидролизу до меперидиновой кислоты [156, 157]. С мочой выделяется менее 5% введенной дозы препарата. Нормеперидин также подвержен гидролизу с образованием нормеперидиновой кислоты. Кислотные метаболиты не обладают биологической активностью и выделяются с мочой в неизмененном и частично в конъюгированном виде [158].
Выделение меперидина с мочой зависит от показателя рН. Если рН мочи опускается ниже 5,0, то с ней выделяется в неизмененном виде около 25% принятой дозы опиоида. Скорость выведения меперидина можно увеличить, способствуя подкислению мочи [159].
Время удаления меперидина колеблется от15 до 40 ч, препарат можно обнаружить в моче даже через 3 дня после его приема. Нормеперидин оказывает возбуждающее действие на ЦНС, его токсические эффекты выражаются миоклонусом и судорогами [157, 160, 161]. Поэтому назначение меперидина больным с почечной недостаточностью может привести к его накоплению и
развитию нормеперидиновой интоксикации [160, 162]. Цирроз печени иногда становится причиной сниженного клиренса и длительной задержки нормеперидина в крови. В то же время больные с циррозом печени до некоторой степени защищены от интоксикации нормеперидином вследствие снижения метаболизма препарата. При повторных назначениях препарата опасность токсических реакций возрастает [163].
Фармакологическое действие. Меперидин почти в 10 раз слабее морфина при энтеральном приеме и в 7-10 раз - при парентеральном введении. В
аналгезирующих дозах не оказывает видимого влияния на -сердечн сосудистую систему. Меперидин в противоположность морфину и другим опиоидам не замедляет ритма сердца. Напротив, благодаря своему структурному сходству с атропином(см. рис. 8-10) он способен спровоцировать тахикардию. В больших дозах меперидин снижает сократительную - спо собность миокарда, величину ударного объема и одновременно повышает давление наполнения. Отрицательный инотропный эффект меперидина проявляется при его назначении в дозе 2-2,5 мг/кг [89, 90].
Незначительное антиспастическое действие этого препарата было -от мечено уже при первом описании свойств этого вещества Eisleb и Shaumann в 1939 г. При назначении в эквивалентных с морфином аналгезических дозах не происходит столь значительного спазма желчных путей[164]. Слабое влияние на гладкую мускулатуру делает меперидин препаратом выбора среди других опиоидов при лечении больных с почечной коликой.
В отличие от других опиоидов меперидин чаще вызывает мидриаз, а не миоз, что отражает его атропиноподобные свойства.
Использование в клинике и фармацевтические препараты. Мепе-
ридин назначают внутримышечно при сильных болях по 75-100 мг [165, 166]. Повторные введения препарата могут потребоваться каждые2-4 ч, так как продолжительность создаваемой им аналгезии меньше, чем у морфина. При ведении послеоперационных больных меперидин назначают в инфузиях, нагрузочная доза составляет 0,5-1,5 мг/кг. Спустя 30-60 мин переходят на поддерживающие дозы 0,25/0,75 мг/мин [167]. Поддерживающие дозы необходимо часто корригировать (см. гл. 10). Меперидин чаще других препаратов используют для ВВ-АКП [138, 168, 169].
Меперидина гидрохлорид выпускают в таблетках по50 и 100 мг, а также в растворах(50 мг в чайной ложке). Препарат для парентерального применения выпускается в различных концентрациях.
Фентанил
Фентанил является производным меперидина и входит в группу фенилпиперидина (см. рис. 8-18). Обезболивающее действие препарата в 75-125 раз сильнее, чем у морфина [170].
Фармакокинетика. Более высокая, чем у морфина, растворимость в липидах объясняет быстрое наступление эффекта после введения фентанила (в течение 30 с) и небольшую продолжительность действия. Высокая липо-
фильность объясняет быстрое и значительное распределение препарата в тканях. В хорошо перфузируемых тканях высокая концентрация фентанила достигается быстро. Эффект от действия фентанила вскоре прекращается в связи с быстрым высвобождением его из жировой ткани и из скелетных мышц и соответственно снижением его содержания в плазме крови [171].
Таким образом, кратковременность действия однократной дозы фентанила отражает быстрое его потребление тканями и столь же быстрое высвобождение с падением уровня препарата в крови. При повторных введениях или при непрерывной инфузии фентанила может наступить насыщение им неактивных жировых и мышечных депо. В этом случае темпы снижения его концентрации в крови замедляются и действие фентанила удлиняется. Следовательно, снижение уровня препарата в крови отражает его удаление, а не распределение в тканях [172].
Метаболизм фентанила проходит путем деалкилирования, гидроксилирования и амидного гидролиза с образованием норфентанила и деспропионилнорфентанила, которые выводятся с мочой и желчью. В неизмененном виде с мочой выделяется не более8% принятой дозы препарата. В противоположность меперидину норметаболиты фентанила неактивны и не оказывают стимулирующего влияния на ЦНС. Считается, что фентанил является препаратом выбора для больных с нарушением функции почек [173, 174].
Несмотря на кратковременность действия фентанила, выведение его из организма происходит относительно медленно. Время полувыведения составляет 185-219 мин, что отражает большой объем распределения препарата в тканях (см. табл. 8-7). Последнее обстоятельство связано с высокой растворимостью препарата в липидах. Цирроз печени не оказывает заметного влияния на сроки выведения фентанила [175]. У лиц пожилого возраста удаление препарата замедлено еще больше, что связано с более медленным клиренсом. Объем распределения препарата у лиц пожилого возраста остается таким же, как и у молодых[176, 177]. Следовательно, в пожилом возрасте действие фентанила может пролонгироваться.
Фармакологическое действие. Фентанил влияет на ЦНС как депрессант, вызывая аналгезию и подавляя дыхание. Весьма примечательно, что в отличие от меперидина фентанил в малых дозах (1-2 мкг/кг) обладает слабым гипнотическим и седативным действием(см. табл. 8-9). Большие дозы, не применяемые в практике обезболивания (50-150 мкг/кг), вызывают глубокую седацию вплоть до потери сознания.
Фентанил при парентеральном введении почти в 100 раз активнее морфина, но, несмотря на это, при введении названных средств в эквивалентных дозах угнетение дыхания развивается в одинаковой степени.
В отличие от морфина фентанил даже в больших дозах не высвобождает гистамин [95]. Введение фентанила способно индуцировать брадикардию,
но выраженной степени она достигает только при анестезирующих дозах препарата [178].
Использование в клинике и фармацевтические препараты. Фента-
нила цитрат применяют в концентрации50 мкг/мл. Препарат выпускают в таблетках вместе с дроперидолом(инновар), однако эта комбинация малопригодна для устранения послеоперационной боли.
Продолжаются интенсивные исследования по внутривенному(см. гл. 10), эпидуральному и субарахноидальному введению фентанила (гл. 11 и 12). Сведений о фармакокинетике фентанила после его внутримышечного введения в литературе нет.
В недавнем прошлом фентанил не назначали энтерально из-за выраженного метаболизма в печени и слабой усвояемости в биологически активной форме (32%) [79]. Однако созданы новые модификации препарата, на-
пример, в виде трансмукозального фентанила цитрата для энтерального употребления, и его усвояемость повышается до 52% [179]. Препарат обычно применяют для предоперационной седации в педиатрии[180, 181] и для купирования болей у больных раком[182, 183]. Дозы варьируют от 10 до 25 мкг/кг. Побочное действие при таких дозах проявляется легким зудом в области лица (65-85%) и слабым зудом всего тела(10-30%) либо выраженной тошнотой (30-37%) [183-186]. Трансмукозальный фентанила цитрат не используют для преодоления послеоперационных болей. Чаще его применяют как дополнительное средство для обезболивания и седации в палатах интенсивной терапии [186]. Однако в последнее время описаны показания для интраназального применения фентанила с целью снятия болей в послеоперационном периоде [187].
Трансдермальное введение фентанила. Разработка трансдермальных методов введения (транстермальные терапевтические системы-ТТС) эстрогенов, клофелина и скополамина вызвало интерес к подобному же методу назначения липофильных опиоидов[188]. Липофильность фентанила, возможность седативного действия и влияние на сердечно-сосудистую систему - все это делает весьма привлекательным трансдермальный путь его введения.
Разработаны четыре системы для эффективного трансдермального введения препаратов. В отношении фентанила используют ТТС, основанную на принципе мембранного проникновения(рис. 8-22). Фентанил помещают в мелкий резервуар из непроницаемой пленки, верхнюю поверхность которого прикрывают мелкопористой мембраной, ограничивающей скорость трансдермального проникновения. Плотный контакт с кожей обеспечивается специальным адгезивным полимером, нанесенным на внешнюю сторону микропористой мембраны. В резервуаре содержится небольшое количество фентанила (до 10 мг) в гелевом матриксе, который и определяет скорость диффузии. При необходимости повысить дозировку достаточно увеличить поверхность контактирующей с кожей ТТС так, чтобы поддерживалось постоянное
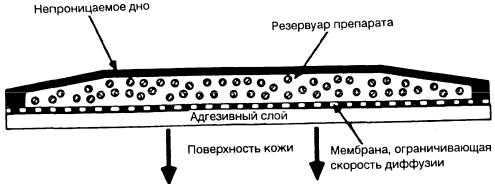
поступление фентанила со скоростью 25, 50, 75 или 100 мкг/ч в течение до 3
дней [188].
Основным препятствием для поступления препарата является роговой слой эпидермиса, поскольку диффузия происходит в основном с участием внутриклеточных липидных сред [190]. Кожа представляет собой как бы резервуар, который должен наполниться, прежде чем будет поддерживаться постоянная абсорбция. В последующем поступление фентанила будет продолжаться даже после удаления ТТС [188].
После наложения ТТС концентрация фентанила в плазме крови возрастает на протяжении 12-18 ч, пока не стабилизируется на определенном уровне (плато). Это состояние соответствует окончательному формированию депо препарата в коже. Концентрация фентанила в плазме крови остается постоянной весь срок прикрепления ТТС к коже. После удаления ТТС уровень препарата в крови постепенно снижается. Периодполу выведения составляет 15-21 ч, что соответствует опорожнению фентанилового депо [190-192].
Рис 8-22 Мембранная пермеация (проникновение) при трансдермальном терапевтическом введении опиоидов. (Из Tarver и Stanley [189], с разрешения.)
Таблица 8-12. Преимущества и недостатки фентанил-ТТС при ведении больных с послеоперационными болями
Преимущества |
Недостатки |
Снижение метаболизма первого |
Невозможность титрования дозы в соот- |
прохождения |
ветствии с повышением или снижением |
|
потребности в аналгсзии |
Стабильная концентрация в кро- |
Заранее выбранная доза |
ви Простота применения |
Медленное наступление действия |
Доза рассчитана на несколько |
Необходимость дополнительной аналге- |
дней |
зии |
Не нужны иглы и инъекции |
Остаточное депо после удаления ТТС |
Эффективная техника аналгезии |
Потенциальная наркотическая зависи- |
|
мость |
По данным всех исследований, посвященных послеоперационной боли, при использовании фентанил-ТТС потребность в опиоидах снижается. Побочные реакции проявляются тошнотой и рвотой(30-85%) или угнетением дыхания [191-196].
Преимущества и недостатки фентанил-ТТС приведены в табл. 8-12. Основной недостаток этой методики связан с титрованием дозы препарата. Эффективное преодоление острой боли зависит именно от возможности варьировать дозировку в соответствии со степенью выраженности болевых ощущений. К сожалению, фентанил-ТСС имеет такие же недостатки, как и другие системы фиксированного дозирования, делающие их малоприемлемыми для преодоления послеоперационной боли(в нашем понимании этой проблемы). Наиболее целесообразно применять фентанил-ТТС для снятия болей у онкологических больных. Трансдермальное назначение фентанила может стать эффективным промежуточным методом на этапе перехода от орального к парентеральному введению опиоидов.
Суфентанил
Суфентанил (см. рис. 8-19) является тиаминовым аналогом фентанила и входит в группу фенилпиперидиповых синтетических опиоидов. Он активнее фентанила в 5-10 раз, соответственно выше и аффинитет его рецепторного связывания.
Фармакокинетика. Высокая растворимость суфентанила в липидах (разделительный коэффициент 1250) согласуется с быстрым проникновением препарата через гематоэнцефалический барьер и объясняет быстрое наступление его действия. Высокий тканевый аффинитет(обусловленный липофильностью препарата) приводит к быстрому его распределению в организме. Как и у фентанила, быстрое перераспределение суфентанила в неактивные ткани (жир, скелетные мышцы) резко ограничивает его действие, особенно при назначении небольших доз [198].
Суфентанил подвержен быстрому метаболизму N-путем деалкилирования пиперидинового азота и 0-деметилирования [199]. Продукты деалкилирования не обладают биологической активностью. Десметилсуфентанил, образующийся при деметилировании, сохраняет примерно 10% активности суфентанила. С мочой выводится менее1% неизмененного суфентанила.
Метаболиты суфентанила выделяются как с мочой, так и с калом. Около 30% выделяющихся метаболитов конъюгируют, преимущественно это происходит с десметилсуфентанилом. Вследствие значительной способности к конъюгации и продукции активных метаболитов назначать препараты -па циентам с почечными заболеваниями следует с осторожностью [200, 201].
Объем распределения суфентанила несколько меньше, а скорость выделения вдвое меньше, чем у фептанила. Суфентанил интенсивно связывается с белками плазмы(90% препарата). Клиренс препарата ослабевает у лиц пожилого возраста, однако время полувыведения не изменяется из-за уменьшения объема перераспределения. Тем не менее у пожилых пациентов может наблюдаться и пролонгированное действие препарата [202].
Фармакологическое действие. Клинико-фармакологический профиль суфентанила почти такой же. как у фентанила, но более выражено седативное действие. При назначении суфентанила несколько чаще развиваются брадикардия, миоз, yгнетение дыхания, тошнота, рвота и спазм гладких мышц.
Применение в клинике и фармацевтические препараты. Для инъ-
екций выпускают суфентапил цитрат в концентрации50 мкг/мл. Для энтерального приема препарат не производят, продолжается его изучение с целью трапсдсрмального назначения [203, 204].
Опыт применения суфентанила для купирования послеоперационной боли весьма ограничен. Основные исследования проводятся по применению суфентанила при анестезии, контролируемой пациентом (АКН), и при эпидуральной анестезии [205 208].
Алфентанил
Алфентанил (см. рис. 8-20) является производным фептанила. Он слабее последнего в 5-10 раз, а продолжительность его действия на1/3 короче, чем у фентанила.
Фармакокинетика. Главными фармакологическими особенностями алфентанила, объясняющими его действие в клинике, служат его низкий рН (6,5) и малый объем распределения [209-211]. Аналгезия после внутривенного введения алфентанила наступает очень быстро(через 1-2 мин). Это может быть связано с низкими величинами рКа, поскольку почти 90% препарата ионизируется при рН 7,4 (см. табл. 8-7). Благодаря высокой ионизации пре-
парат быстро проникает через гематоэнцефалический барьер несмотря на слабую растворимость в липидах.
Объем распределения алфентапила в4-5 раз меньше, чем у фентанила [209, 210]. Слабый тканевый аффинитет алфентанила отражает его слабую растворимость в липидах и высокую степень связывания с протеинами.
Небольшая продолжительность действия алфентанила обусловлена его перераспределением в неактивные ткани и столь же быстрым метаболизмом в печени, как и у суфентанила. С мочой выделяется менее 1% неизмененного алфентанила.
Время полувыведения препарата составляет70-98 мин [210]. У больных с циррозом печени этот показатель возрастает до219 мин [211]. Кроме того, возрастание свободной фракции алфентанила у больных с печеночной патологией объясняется изменением состава белков крови и нарушением их способности связывать препарат. Более медленное выделение алфентанила и увеличение его свободной фракции у больных с циррозом печени могут привести к усилению и к удлинению действия препарата. У больных с патологией почек клиренс алфентанила не нарушается, но объем распределения может измениться в зависимости от связывания препарата белками плазмы. Та-

ким образом, выделение алфентанила не нарушается при патологии почек, но нарушение связывания с белками способно влиять на его распределение.
Использование в клинике и фармацевтические препараты. Подоб-
но суфентанилу, лучше всего изучено применение алфентанила для эпидуральной анестезии [213, 214] и для внутривенной анестезии, контролируемой пациентом (АКП) [213-216].
Краткость действия алфентанила имеет значение для его использования при АКП. В то же время краткосрочность действия может потребовать чрезвычайно больших дозировок и приведет к истощению собственных возможностей пациента в процессе АКП[216]. К сожалению, проведение инфузий, ориентированных на нижнюю границу потребностей пациента, не обеспечивает достаточного обезболивания [215, 216].
В настоящее время алфентанил выпускают только для инъекций, концентрация раствора 500 мкг/мл.
Метадон (дольфин)
Метадон впервые был синтезирован немецкими фармацевтами в период второй мировой войны. Двойная пространственная формула метадона несколько напоминает морфин(рис. 8-23). Однако объемные характеристики придают метадону конфигурацию, более сходную с псевдопиперидиновым кольцом. (Структура пиперидинового кольца, по-видимому, является обязательным условием опиоидной активности.) Левовращающий изомер активнее правовращающего в 8-50 раз, что придает ему основное значение в обезболивании [72].
Рис. 8-23. Метадон.
Фармакокинетика. Метадон хорошо всасывается при энтеральном применении (41-90%) [217, 218]. В плазме крови препарат обнаруживают уже через 30 мин после приема, а пик его концентрации наступает через 4 ч. Признаки аналгезии появляются через30-60 мин после проглатывания. Достаточная концентрация в крови после подкожного введения достигается через 10 мин, а пик концентрации в мозговой ткани - через 1-2 ч.
Метадон подвергается интенсивной биотрансформации в печени, в основном путем N-деметилирования и циклизации с формированием пироли-
динов и пирролина. Неактивные метаболиты вместе с небольшим количеством неизмененного метадона выводятся с мочой и желчью[220, 221]. Под-
кисливание мочи способствует повышению выделения метадона с мочой
[222].
Метадон прочно связывается белками крови и тканей, поэтому при повторных его назначениях может накапливаться в тканях. Постепенное высвобождение метадона из этих соединений поддерживает его содержание в крови и объясняет замедленный клиренс препарата[221, 223] (см. табл. 8-7). Время полувыведения метадона после однократного внутривенного введения составляет 14 ч, а половина оставшейся дозы выводится уже за55 ч по мере истощения тканевых запасов препарата. При систематическом приеме метадона время его полувыведения равно 22 ч [221].
Фармакологическое действие. Профиль клинико-фармакологического действия метадона такой же, как у морфина. Несмотря на длительное время первичного полувыведения (14 ч), продолжительность аналгезии не превышает 4-5 ч [224]. При повторных приемах наблюдается кумулятивный эффект (высвобождение метадона из тканей). В случае длительного энтерального приема последующие дозировки препарата следует снижать или увеличивать интервалы между приемами. Действенность энтерального применения метадона к парентеральному соотносится как 1:2.
Использование в клинике и фармацевтические препараты. Мета-
дон считается отличным анальгетиком, наиболее пригодным для устранения хронических болей [225, 226]. Некоторые работы посвящены его эпидуральному назначению [227, 228]. В то же время исследований по егоис пользованию для внутривенной АКП не проводилось. Правда, в серии исследований Gourlay и сотр. [223, 229, 230] продемонстрирована высокая эффективность интермиттирующих внутривенных введений метадона для устранения послеоперационных болей.
Метадона гидрохлорид выпускают для энтерального употребления в таблетках по 5 и 10 мг и в растворе(10 мл в чайной ложке). Для инъекций производят растворы с концентрацией 10 мг/мл.
Пропоксифен (дарвоцет, вигезиг)
Пропоксифен (рис. 8-24) по своей структуре очень близок к метадону, но обладает более слабым аналгезирующим действием и применяется только энтерально. Известны четыре изомера препарата, обладающие только -а анальгегической активностью. В отличие от метадона обезболивающее действие оказывает только правовращающий изомер [72].
Фармакокинетика. Пропоксифен подвергается интенсивному метаболизму при первом прохождении, поэтому его биологическое действие при энтеральном пути введения проявляется на30-70%. Процессы метаболизма
состоят в деметилировании с образованием норпропоксифена, который постепенно выделяется с мочой. Время полувыведения норпропоксифена составляет примерно 23 ч, а самого пропоксифена - 14,6 ч. Поэтому при повторных приемах препарата уровень норпропоксифена в крови может -ока заться в 4 раза выше, чем самого пропоксифена [231].
Рис. 8-24. Пропоксифен.
Использование в клинике и фармацевтические препараты. В клинике пропоксифен используют как легкий анальгетик и назначают только энтерально. Его доза 90-120 мг эквивалентна 60 мг кодеина или 600 мг аспирина [232]. Пропоксифен не оказывает противовоспалительного и жаропонижающего действия, его противокашлевое влияние незначительно.
Пропоксифена гидрохлорид выпускают в таблетках по32 и 65 мг, а пропоксифена напсилат - в таблетках по 100 мг или в виде суспензии.
ОПИОИДНЫЕ АГОНИСТ-АНТАГОНИСТЫ
Опиоиды со свойствами агонист-антагонистов могут быть разделены на две группы по принципу их истинной активности(см. табл. 8-5). Смешанные агонист-антагонисты действуют одновременно как агонисты на одни рецепторы и как антагонистына другие. (Сам термин «смешанные агонистантагонисты» иногда по ошибке используют обобщенно, включая в него обе группы этих соединений.) К смешанным агонист-антагонистам
Таблица 8-13. Клинические свойства опиоидных агонист-антагонистов по сравнению с чистыми агонистами
Сниженная способность к аналгезии по сравнению с чистыми μ-агонистами Выраженный «потолочный» эффект как в отношении аналгезии, так и угнетения дыхания Антагонистические свойства могут быть использованы для снятия действия
μ-агонистов (угнетение дыхания, зуд, тошнота, рвота) при сохранении аналгезии Дисфорическая реакция

Малая вероятность наркотической зависимости относятся налбуфин, пентазоцин и буторфанол.
Частичные агонисты (бупренорфин, дезоцин) оказывают субмаксимальное действие только на рецепторы определенного типа (см. рис. 8-5).
Клинические проявления действия препаратов обеих групп весьма сходны
(табл. 8-13).
Смешанные агонист-антагонисты
Налбуфин (нубаин)
Налбуфин (рис. 8-25) является полусинтетическим смешанным аго- нист-антагонистом, близким по своей химической структуре оксиморфону и налоксону [75].
Фармакокинетика. После приема внутрь налбуфин подвергается интенсивному метаболизму при первом прохождении в печени. Биологическое действие оказывает около10% принятой дозы препарата[233]. Основным продуктом метаболизма является неактивный глюкуронид. Выведение налбуфина и его метаболитов происходит в основном с калом. Только 7% введенной дозы выделяется с мочой в виде неизмененного препарата или его метаболитов. Время полувыведения налбуфина 36 ч [234].
Фармакологическое действие. Как парциальный агонист налбуфин воздействует прежде всего на капа-рецепторы. Его аналгезирующие свойства связаны именно с этим рецепторным воздействием. Налбуфин одновременно является антагонистом в отношении μ-рецепторов [235].
Рис. 8-25. Смешанные агонист-антагонисты.
При внутримышечном введении налбуфин вызывает обезболивание, вполне сравнимое с действием морфина(соотношение действия 1:1). Время наступления обезболивания и его продолжительность такие же, как и у морфина. При повышении дозы сверх0,45 мг/кг обезболивающее действие не усиливается, угнетение дыхания не усугубляется [236].
Аналгезирующее действие μ-агонистов, введенных после налбуфина, заметно ослабевает. Этот препарат также способен ускорять развитие симптомов отмены у лиц с физической зависимостью от опиоидов. Однако μ- антагонистические способности налбуфина можно использовать для быстрого восстановления дыхания, подавленного μ-агонистами. При этом обезболивающее действие последних сохранится. Точно так же налбуфин можно ис-
пользовать для противодействия другим неблагоприятным влияниям- μ агонистов, например для устранения зуда [237-240].
Седативный эффект относится к наиболее частым проявлениям побочного действия налбуфина, наблюдающегося у 33% пациентов. Нередко имеет место и обильное потоотделение. Дисфория наблюдается реже, чем при назначении пентазоцина или буторфанола, однако частота ее увеличивается по мере нарастания дозировки. В отличие от пентазоцина и буторфанола налбуфин не повышаег артериальное давление, давление в системе легочной артерии и частоту сердечных сокращений [241].
Использование в клинике и фармацевтические препараты. Как бы-
ло отмечено выше, налбуфин весьма эффективен в реверсии побочного действия μ-агонистов, сохраняя в то же время их обезболивающее влияние. Опыт применения налбуфина для ВВ-АКП и для эпидуральной аналгезии невелик [242-245]. Можно полагать, что налбуфин весьма эффективен при ВВАКП, но слабо действует при эпидуральной введении.
Обычная доза препарата при внутримышечном или внутривенном введении взрослым равна 10 мг каждые 3-6 ч. Максимальная суточная доза составляет 160 мг. Налбуфина гидрохлорид выпускают в виде раствора для инъекций (10 мг/мл).
Пентазоцин (тальвин)
Пентазоцин является синтетическим опиоидом из группы бензоморфана (см. рис. 8-25). Аналгезирующие его способности связаны с левовращающим изомером этого препарата.
Фармакокинетика. Пентазоцин хорошо абсорбируется при энтеральном и парентеральном введении. После приема внутрь биологическое действие оказывает не более20% введенной дозы препарата из-за интенсивного его метаболизма в печени[246]. Метаболизм происходит в основном путем оксидации концевых метальных групп с образованием неактивных глюкуро-
нидов. Последние выделяются с мочой вместе с 5-25% неизмененного препа-
рата [247].
Фармакологическое действие. Пентазоцин действует как антагонист на капа-рецепторы (вызывая аналгезию) и на дельта-рецепторы(вызывая психомиметический и дисфорический эффект). При парентеральном введении пентазоцин в 2-3 раза слабее морфина. Препарат также вызывает седативное действие (капа-агонистический эффект) и потоотделение. Высокие дозы пентазоцина провоцируют галлюцинации и психомиметический эффект («активная тревога», деперсонализация, ощущение «надвигающейся судь-
бы») [248].
Пентазоцин вызывает такое же угнетение дыхания, как и эквивалентные дозы μ-агонистов. Среди смешанных агонист-антагонистов он обладает максимальной способностью подавлять дыхание и вызывать аналгезию. При дозе препарата выше 30 мг пропорционального возрастания дыхательной депрессии не происходит. У взрослого человека с массой тела 70 кг максимальный эффект наступает при дозе 60 мг [249, 250].
Пентазоцин не вызывает гипотензии и брадикардии, однако может привести к повышению кровяного давления и давления в малом круге, учащению пульса. Влияние пентазоцина на давление в желчных путях выражено слабее, чем у других μ-агонистов [251-252].
Опасность привыкания невелика, но все же иногда появляются физическая зависимость и наркомания. Пентазоцин способен ускорять развитие синдрома отмены у лиц с физической зависимостью от опиоидов.
Использование в клинике и фармацевтические препараты. Пента-
зоцина лактат выпускают в растворе для инъекций концентрацией30 мг/мл. Для приема внутрь выпускают пентазоцина гидрохлорид с ацетилсалициловой кислотой (12,5 мг пентазоцина и 325 мг ацетилсалициловой кислоты) или с налоксоном (тальвин NX: 50 мг пентазоцина гидрохлорида и 0,5 мг налоксона).
Доза пентазоцина для взрослых-30 мг внутримышечно или 50 мг каждые 3-4 ч энтерально. В дозе 50 мг пентазоцин оказывает анальгетическое действие, эквивалентное 60 мг кодеина [72]. Назначение пентазоцина для эпидуральной анестезии и для ВВ-АКП изучено недостаточно.
Буторфанол (стадол)
Буторфанол представляет собой синтетический смешанный агонистантагонист морфинового ряда. Агонистическое его действие выражено в20 раз, а антагонистическое в 10-30 раз сильнее, чем у пентазоцина [253].
Фармакокинетика. Сведения о фармакокинетике буторфанола весьма неполные. Препарат быстро и полностью абсорбируется после внутримышечного введения. Уже спустя 5 мин распределяется
1/2 введенного препарата. Окончательное время полураспределения, учитывая высокий клиренс препарата, составляет 160 мин [254, 255].
Впроцессе метаболизма буторфанола образуется гидроксибуторфанол
инорбуторфанол. Оба метаболита неактивны. Почки выделяют около 25% введенной дозы препарата, и около 85% его количества связывается с белками плазмы крови [255].
При энтеральном приеме буторфанол малоэффективен из-за интенсивного метаболизма при первом прохождении в печени.
Фармакологическое действие. Буторфанол обладает слабым аффинитетом в отношении μ-рецепторов(антагонист), выраженным аффинитетом в отношении капа-рецепторов (вызывает аналгезию) и минимальным - к дель- та-рецепторам (слабый дисфорический эффект). Буторфанол в 3-5 раз более действен в отношении аналгезии, чем морфин.
Подобно другим смешанным агонист-антагонистам, буторфанол вызывает седацию и диафорез. Он может подавлять дыхание и вызывать аналгезию, как это свойственно смешанным агонист-антагонистам. Подобно пентазоцину, буторфанол повышает общее артериальное давление. давление в легочной артерии и частоту сердечных сокращений[241, 256]. Влияние его на желчные пути выражено слабее, чем у μ-агонистов [89].
Обезболивающее действие μ-агонистов может оказаться малоэффективным, если пациенты перед этим получали буторфанол. Данный препарат не устраняет депрессию дыхания, как это происходит при назначении налбуфина, но другие побочные реакции на μ-агонисты(например, зуд) после парентерального введения буторфанола исчезают [257].
Использование в клинике и фармацевтические препараты. Показа-
нием для назначения буторфанола служат выраженные или сильные боли. Внутримышечно препарат рекомендовано вводить в дозах12 мг, а внутривенно по 0,5-2 мг каждые 3-4 ч. Буторфанол выпускают только для паренерального применения в растворе с концентрацией 1 или 2 мг/мл.
Буторфанол хорошо зарекомендовал себя как средство для эпидуральной анестезии, в частности при кесаревом сечении[258-260]. Хорошее обезболивание обеспечивается при введении 2 мг препарата в 10 мл нормального солевого раствора без консервантов. Эпидуральное введение препарата дает также и седативный эффект, особенно при повышенных дозировках [258].
Частичные агонисты Бупренорфин (бупренекс)
Бупренорфин - это полусинтетический опиоид, производное опиоида тебаина (рис. 8-26).
Фармакокинетика. Бупренорфин обладает высокой липофильностью [261]. В отношении этого препарата подтверждается ранее высказанное в

этой главе положение, что кинетика обезболивающего действия анальгетика после его парентерального введения регулируется не общей фармакокинетикой, а преимущественно динамикой рецепторных диссоциаций[41, 44]. Диссоциация бупренорфина с μ-рецепторами происходит замедленно, и это объясняет продолжительность действия препарата, не совпадающую с временем его полувыведения из крови, составляющим 3-5 ч [262, 263]. Следовательно, прямой связи между содержанием бупренорфина в крови и его фармакологическим действием нет. Примерно 96% введенной дозы препарата связывается белками сыворотки [263].
Рис. 8-26. Бупренорфин.
Фармакологическое действие. Бупренорфин - высокоактивный препарат. При внутривенном его введении0,3 мг этого препарата эквивалентны
10 мг морфина. На μ-рецепторы он действует как частичный агонист. Однако его аффинитет к этим рецепторам примерно в50 раз сильнее, чем у морфина
[264].
Побочные реакции на бупренорфин напоминают таковые у смешанных агонист-антагонистов. Седация и сонливость отмечаются почти 50%,у а тошнота и рвота - у 10-20% пациентов. Бупренорфин не вызывает психомиметических и дисфорических реакций, поскольку не является агонистом в отношении дельта-рецепторов.
Отмечено выраженное угнетение дыхания под влиянием бупренорфина. Это воздействие может быть весьма продолжительным из-за значительного и длительного аффинитета препарата к μ-рецепторам [265, 266]. По этой же причине реверсия подобного действия бупренорфина достигается лишь при назначении высоких доз налоксона[267]. Для поддержания адекватной вентиляции у больных, получивших большие дозы бупренорфина, можно применять доксапрам [268]. Обладая свойствами парциального агониста, бупренорфин способен несколько ослаблять действие высоких доз других- μ агонистов, уменьшая тем самым угнетение дыхания, вызванное этими опио-
идами [269].
На сердечно-сосудистую систему бупренорфин оказывает такое же воздействие, как и морфин.
Использование в клинике и фармацевтические препараты. Бупре-
норфин обычно назначают внутримышечно или внутривенно в дозе0,3 мг каждые 6 ч. При этом наиболее эффективно устраняются выраженные и тя-

желые боли в послеоперационном периоде, болевой синдром при почечной колике, раке, инфаркте миокарда [270, 271].
ВЕвропе бупренорфин выпускают для сублингвального употребления
вдозах 0,4-0,8 мг. Этот путь введения обеспечивает хорошую послеопераци-
онную аналгезию. Препарат также используется для АКП в - |
посл |
операционном периоде [272]. |
|
Для проведения ВВ-АКП бупренорфин теоретически является непод- |
|
ходящим препаратом. Аналгезия наступает медленно, но, развившись, про- |
|
должается длительно благодаря высокому аффинитету препарата- |
к μ |
рецепторам. По этой причине на фоне повторных назначений препарата мо- |
|
жет наступить передозировка, сопровождающаяся дальнейшим ухудшением |
|
дыхательной функции [273]. Однако были сообщения [274, 275] о том, что |
|
при проведении ВВ-АКТ бупренорфин обеспечивал хорошую аналгезию при |
|
минимальном угнетении дыхания. |
|
Эпидуральное введение бупренорфина применяют для обезболивания |
|
после кесарева сечения и ортопедических операций[261, 276-278]. Бупре- |
|
норфин, подобно налбуфину и буторфанолу, используют для устранения зуда |
|
после эпидурального введения μ-агонистов[279]. Субарахноидальному введению бупренорфина для аналгезии после операции кесарева сечения было посвящено всего одно исследование [280].
Бупренорфин выпускают в растворе концентрацией 0,3 мг/мл.
Дезоцин (далган)
Дезоцин относится к синтетическим препаратам морфинного ряда и обладает выраженными свойствами парциального μ-агониста (рис. 8-27).
Рис. 8-27. Дезоцин.
Фармакокинетика. Опубликованы результаты всего одного исследования по фармакокинетике дезоцина у человека[280]. Препарат быстро распределяется в организме, но медленно выводится. Объем распределения у него весьма высок, что указывает на его интенсивное поглощение тканями. Дезоцин выделяется почками в неизмененном виде, а также подвергается метаболизму в печени [281].
Фармакологическое действие. По механизму действия дезоцин относится к частичным μ-агонистам, но одновременно он обладает свойствами дельта-агониста и в минимальной степени - капа-агониста. В отличие от нал-
буфина, пентазоцина и буторфанола дезоцин усиливает обезболивающий эффект даже при его назначении после других μ-агонистов[283]. При парентеральном введении действие дезоцина эквивалентно морфину. Аналгезический эффект дезоцина быстро ревертируется под влиянием налоксона как в эксперименте, так и в клинике [284, 285]. Препарат не так плотно фиксируется μ-рецепторами, как бупренорфин.
Дезоцин вызывает побочные реакции, характерные для агонистантагонистов в целом: седацию (иногда продолжительную), тошноту и рвоту. Седативный эффект наступает через1 ч после внутримышечного введения 10-15 мг дезоцина [286]. Опубликовано сообщение о дисфорических реакциях после введения больших доз дезоцина, хотя аффинитет препарата к дель- та-рецепторам невелик [287].
Угнетение дыхания под влиянием повышенных доз дезоцина имеет характер «потолка» действия [283, 284]. При дозах свыше 0,3 мг/кг не происходит дальнейшего увеличения ни аналгезии, ни угнетения дыхания [288].
Влияние дезоцина на сердечно-сосудистую систему пока недостаточно изучено. По данным единичных исследований больных, подвергшихся катетеризации сердца, дезоцин в дозе 0,125 мг/кг вызывает непродолжительное повышение давления в легочной артерии и сосудистого сопротивления в малом круге. Частота сердечных сокращений и артериальное давление не изме-
няются [288, 289].
Вероятность развития наркотической зависимости минимальная, хотя имеются сообщения о наркотическом потенциале дезоцина[290, 291]. Возможности дезоцина провоцировать развитие синдрома отмены у пациентов с зависимостью от опиоидов не изучены.
Использование в клинике и фармацевтические препараты. Дезо-
цина лактат выпускают в растворах концентрацией5, 10 и 15 мг/мл. Было проведено несколько клинических исследований по парентеральному применению дезоцина с целью послеоперационной аналгезии [292-295], однако истинная его роль в этом отношении остается неясной. Отсутствует также опыт по использованию этого препарата для АКП или для энидуральной аналгезии.
АНТАГОНИСТЫ
Небольшие изменения химической структуры μ-агонистов способны трансформировать их в антагонисты по отношению к одному или многим рецепторам [75]. Налоксон является N-аллил-(—CH2—CH=CH2) производным оксиморфона (см. рис. 8-15). Налтрекс имеет циклопропилметиловую группу (—СН2—<) на третьем атоме азота. Известны и другие опиоидные антагонисты, и первый из них - налорфин, а также налмефен и холецистокинин[296298]. Они не обсуждаются в данной главе.
Налоксон и налтрексон являются чистыми антагонистами в отношении μ-, капа- и дельта-рецепторов. Оба последних препарата обладают высоким аффинитетом к μ-рецепторам. Их аффинитет к дельта- и капа-рецепторам менее выражен, но тем не менее они вытесняют агонисты из связи с соответ-

ствующими рецепторами. Однако после вытеснения налоксон и налтрексон не активируют опиоидные рецепторы, вызывая антагонистическое действие.
Парентеральные антагонисты Налоксон (наркан)
Фармакокинетика. Внутривенное введение налоксона в дозе1-4 мкг/кг приводит к реверсии аналгезии и дыхательной депрессии, вызванных опиоидами. Продолжительность этого действия невелика(30-45 мин), что, вероятно, связано с метаболизмом и быстрым вымыванием препарата из рецепторов в мозге [299]. Поэтому для поддержания антагонистического действия препарата бывают необходимы повторные его введения.
Налоксон подвергается метаболизму при первом прохождении в печени (рис. 8-28). Время его полувыведения составляет 64 мин [300]. Особенно интенсивный метаболизм наблюдается при энтеральном приеме налоксона.
Рис. 8-28. Налоксон.
Фармакологическое действие. В отношении опиоидов, назначаемых обычными путями (не нейрогенными), налоксон действует как антагонист, устраняя вызванное ими нарушение дыхания и аналгезию (см. табл. 8-6). При специальном подборе дозировок можно сохранить, хотя бы частично, аналгезию при минимально выраженной дыхательной депрессии. Однако тошнота, рвота и стимуляция сердечно-сосудистой деятельности могут сопровождать частичное ослабление аналгезии.
Действие налоксона, введенного эпидурально, возрастает соответственно повышению дозировки. При инфузии налоксона в дозе5 мкг/кг в 1 ч качество вызываемой эпидуральным введением морфина аналгезии не изменяется, но устраняются нарушения дыхания. Инфузия налоксона в дозе10 мкг/кг в 1 ч уже снижает аналгезию[301]. Введение налоксона в дозах5-10 мкг/кг в 1 ч устраняет как депрессию дыхания, так и аналгезию, вызванную фентанилом [302]. Причины столь выраженной разницы между морфином и фентанилом неясны.
Появление тошноты и рвоты непосредственно связано со скоростью введения налоксона [303, 304]. Малые дозы препарата, назначаемые каждые 2-3 мин, снижают частоту этих побочных реакций. Налоксон может увеличивать нагрузку на сердечно-сосудистую систему. Это выражается активацией симпатической нервной системы: тахикардия, гипертензия, аритмия, отек легких [305-307].