

2.3. Формулировка первого начала термодинамики
Первое начало термодинамики – это закон сохранения энергии. Он впервые четко сформулирован Г. Гельмгольцем в 1847 году. Этот закон не может быть четко доказан, но является результатом всего человеческого опыта. Такие законы часто называют законами природы. Известный математик А. Пуанкаре как-то иронически заметил, что в сущности все твердо верят в закон сохранения энергии потому, что математики принимают его за экспериментальный факт, а экспериментаторы считают его математической теоремой.
Есть несколько формулировок первого закона термодинамики. Если одна из них принимается в качестве основной, то все другие являются следствиями, вытекающими из нее.
Одна из формулировок звучит следующим образом: энергия не создается и не уничтожается. Возможны лишь превращения энергии из одного вида в другой в строго эквивалентных количествах.
Следствием из этого закона является вывод, что невозможен вечный двигатель первого рода, т.е. нельзя создать такой двигатель, который совершил бы работу без затраты энергии. Создание такого двигателя возможно только в том случае, если неверен первый закон.
Часто используется еще одна формулировка первого начала термодинамики: внутренняя энергии изолированной системы есть величина постоянная.
Если данной системе передается некоторое количество энергии в форме тепла Q, которое идет только на приращение внутренней энергии системыUи на совершение системой работыW, то, согласно первому началу,
Q=U+W, (2.1)
для бесконечно малых изменений
Q=dU+W. (2.2)
Уравнения (2.1) и (2.2) являются математическим выражением первого начала термодинамики.
Укажем, что UиdUне зависят от пути перехода системы из начального состояния в конечное, т.е. внутренняя энергия является функцией состояния системы.
Справедливость этого утверждения можно доказать следующим образом (рис. 2.1).
Предположим, что в состоянии (I) внутренняя энергия системы U1. Из этого состояния система переходит в состояние (2), в котором ее внутренняя энергия равнаU2.
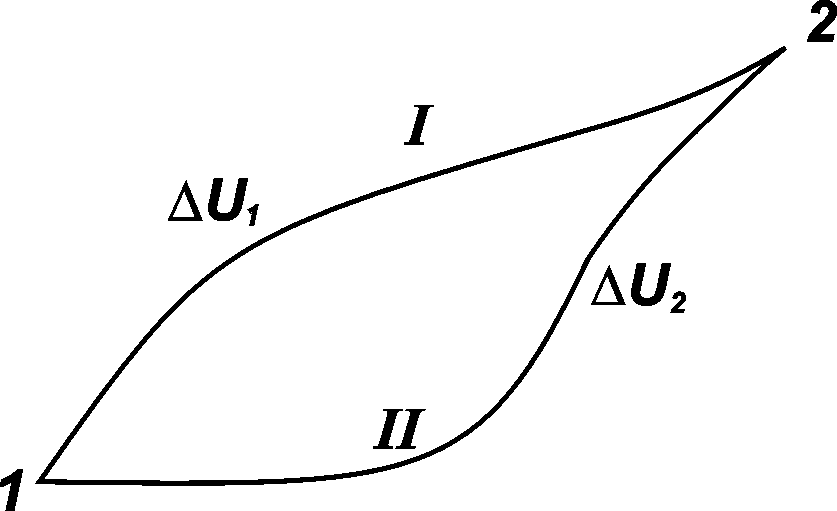
Рис. 2.1. Схематическое отображение путей перехода системы из состояния 1 в состояние 2 и обратно
При переходе системы из состояния 1в состояние2 по путиIобозначим изменение внутренней энергии системы какU1, а по второму пути, – какU2. Согласно первому закону,U1 = U2. Если бы это равенство не соблюдалось, а, например,U1было бы больше U2, то, переводя систему из состояния 1 в состояние 2 по пути I и обратно по пути II, можно было бы получать энергию из ничего. Это противоречит первому началу термодинамики.
В отличие от UвеличиныQиWв общем случае зависят от пути процесса. Поэтому в уравнении (2.2)dUявляется полным дифференциалом, в то время какQиW– просто бесконечно малыми величинами
2.4. Применение первого начала термодинамики к различным процессам
Если система совершает работу Wтолько против внешнего давления, то
W = РV (2.3)
или для элементарного процесса
W = pdV. (2.4)
Математическое выражение первого начала термодинамики в этом случае имеет вид:
Q= U + p (2.5)
или
Q = dU + pdV. (2.6)
Для изохорного процесса (V = const) pV = 0и следовательно:
QV =U, (2.7)
QV =dU, (2.8)
где QV– теплота, сообщаемая системе в изохорном процессе.
Из уравнений (2.7) и (2.8) следует, что в изохорном процессе тепло, сообщаемое системе, идет только на приращение внутренней энергии системы. В этом случае QV не зависит от пути перехода системы из одного состояния в другое иQVявляется полным дифференциалом, т.е. функцией состояния.
Это означает, что изменение внутренней энергии системы U в изохорном процессе определяется теплотой процессаQVи может быть измерено калориметрически.
В случае изобарного процесса (р= const)
QP =U+PV, (2.9)
Qp =U2 -U1+pV2-pV1, (2.10)
где QP– теплота, сообщаемая системе в изобарном процессе. Сгруппируем величины с одинаковыми индексами
QP= (U2 +PV2) - (U1+PV1). (2.11)
Так как внутренняя энергия, объем системы и давление – функции состояния, то сумма величин U + pVтакже должна быть функцией состояния и ее изменение не зависит от пути перехода системы из одного состояния в другое.
Эту функцию состояния называют энтальпией и обозначают символом Н.
Н = U + pV. (2.12)
Из уравнений (2.11 ) и (2.12) следует, что
Qp=Н, (2.13)
Qр= dH. (2.14)
Таким образом, в изобарном процессе все тепло, сообщенное системе, идет на приращение ее энтальпии. В этом случае Qpтакже не зависит от пути перехода системы из одного состояния в другое иQрявляется полным дифференциалом.
Из уравнений (2.13) и (2.14) следует также, что изменение энтальпии системы Нв изобарном процессе определяется теплотой процессаQpи может быть измерено калориметрически.
В адиабатном процессе, т.е. в процессе, в котором система не обменивается с окружающей средой теплотой, работа совершается за счет уменьшения внутренней энергии системы:
Q= 0 , (2.15)
Q= 0, (2.16)
W= -U, (2.17)
W= -dW. (2.18)
Изотермический процесс характеризуется T = const.для такого процесса dU = 0, тогдаQ = WT, т.е. вся сообщенная системе теплота превращается в работу, величина которой определяется уравнениями
WT=pdv,(2.19)
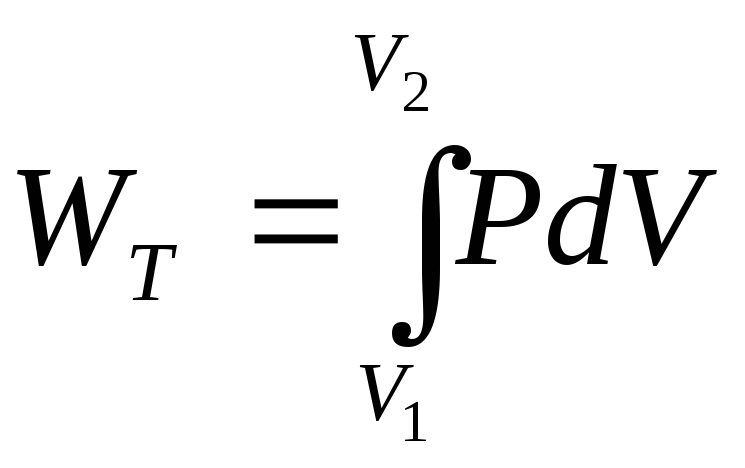 . (2.20)
. (2.20)
Используя уравнение состояния газа Менделеева-Клапейрона, найдем, что для 1 моль идеального газа
![]() ,
,
тогда
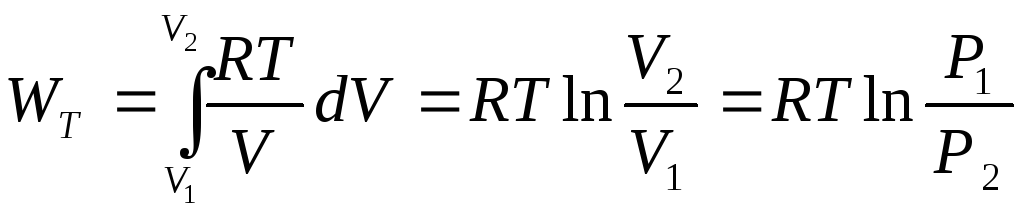 . (2.21)
. (2.21)