

Глава 2. Аномалії хромосом о.Я. Гречаніна, а.В. Христич, т.М. Ткачова
Остеопороз супроводжує вітальні форми хромосомних хвороб насамперед тому, що саме ці варіанти асоційовані з подоланням порога летальності. Наведене спостереження спонукало нас вивчити хромосомні хвороби як причетні до остеопорозу і разом з цим підтвердити нові уявлення про патогенетичні механізми порушень при хромосомних хворобах.
Клінічний випадок.
Скарги:на болі в хребті, колінних, ліктьових, плечових суглобах, скутість рухів, швидку стомлюваність, слабість, відсутність самостійних менструацій, затримка росту і полового розвитку, деформація хребта, грудної клітки.
Анамнез: ДК хворіє з народження, коли батьки виявили набряк нижніх кінцівок, що через 2 тижні пройшов без лікування. Через рік батьки звернули увагу, що дівчинка трохи відстає в рості, хоч психічно розвивається з випередженням. Фізичний розвиток з 11 років характеризувався зниженням зрісто-вагових показників, інтелектуальний розвиток був високим: добре вчилася в школі й університеті. Психоемоційний розвиток також був нормальним. У 12-літньому віці була травма правого гомілковостопного суглобу. Незабаром з'явився біль в лівому гомілковостопному суглобі, що порушувало ходьбу. Лікувалася в НДІ ОЗДП, було встановлено діагноз хронічного артриту, дисгенезії гонад, мозаїчна форма синдрому Шерешевського-Тернера. З перенесених захворювань у дитинстві відзначає гострі респіраторні інфекції, ексудативний діатез на тлі вживання сумішей, травма лівої гомілки і чола. У 1993р. встановлено діагноз ревматоїдного артриту, суглобної форми, повільно прогресуючий хронічний плин, хронічний тонзиліт. З 15 років, у зв'язку з первинною аменореєю, одержувала гормональне лікування мікрофоліном, після чого з'явилися менструалоподібні виділення. В 18 років самостійно припинила гормональне лікування. В 19 років з'явилися зміни з боку хребта, кіфосколіоз, дифузійний остеопороз, контрактури великих суглобів (рис. 1). Сформувалася вторинна кардіоміопатія, пролапс мітрального клапана. У 2004 р. відбувся енергетичний перелом стегна, після чого різко розвилася м'язова гіпотонія, остеопороз, дислокація С4 -С5, підсилилися болі в суглобах і з'явилися обмеження руху в них. Хвора втратила здатність самостійно пересуватися. Генетики ХСМГЦ разом з ортопедами-травматологами проводили реабілітаційні заходи, спрямовані на нормалізацію енергетичного обміну й обміну сполучної тканини, що характеризувалося ураженням проміжного обміну. Запідозрено формування у хворої з мозаїчною формою синдрому Шерешевського-Тернера вторинної мітохондріопатії, порушення проміжного обміну сполучної тканини по типу мукополісахаридоза, тобто захворювання почало носити характер проявів епігенетичної хвороби з проявами фенотипової синтропії.

Рис. 1.
При обстеженні родини пробанду:
Таблиця 1
Соматогенетичне дослідження родини
Особливості фенотипу |
Пробанд |
Мати |
Батько |
Брат (13 років) |
Ріст |
145 |
165 |
175 |
157 |
Невуси на шкірі |
+ |
- |
- |
- |
Телеангіектазії |
+ |
- |
- |
- |
Птерігіум |
+ |
- |
- |
- |
Сухість шкіри |
+ |
- |
- |
- |
Ламкі нігті |
+ |
- |
- |
- |
Нігті у виді годинних стрілок |
+ |
- |
- |
- |
Гіпертрихоз |
+ |
- |
- |
- |
Підшкірна клітковина не змінена |
+ |
+ |
+ |
+ |
Череп не змінений |
+ |
+ |
+ |
+ |
Широке обличчя |
+ |
+ |
- |
- |
Відстовбурчені вушні раковини |
+ |
- |
- |
- |
Деформація вушних раковин |
+ |
- |
- |
- |
Згладжений малюнок вушних раковин |
+ |
- |
- |
- |
Прирослі мочки ушей |
+ |
- |
- |
- |
Епікант |
+ |
- |
- |
- |
Гіпертелоризм |
+ |
- |
- |
- |
Короткий фільтр |
+ |
- |
- |
- |
Відкриті нагору ніздрі |
+ |
- |
- |
- |
Широка спинка носа |
+ |
- |
- |
- |
Макростомія |
+ |
- |
- |
- |
Горизонтальна виступаюча верхня губа |
+ |
- |
- |
- |
Готичне небо |
+ |
- |
- |
- |
Карієс |
+ |
- |
- |
- |
Брахідактилія |
+ |
- |
- |
- |
Вальгусна установка рук |
+ |
- |
- |
- |
Контрактура колінних суглобів |
+ |
- |
- |
- |
При ехографії органів черевної порожнини виявлено переваскулярну інфільтрацію в печінці, надлишок сполучнотканинних структур у перипортальній області, ознаки панкреатопатії, непрямі ознаки дуоденіту.
Нирки - двосторонній нефроптоз, диспластичні і метаболічні зміни, гідрокалікоз. При гінекографії: різко виражена гіпоплазія матки, дисгенетичні яєчники.
Рентгенограма черепа і грудного відділу хребта (22.01.2002р.): у грудному відділі визначається кіфосколіоз Тh6-Т7, у Тh8-тh9 - частковий анкілоз, у сегментах С3-С7 і Тh6-тh10 визначається явище спондилоартрозу й остеохондрозу. На тлі дифузійного остеопорозу відзначається завуалірованність куприко підвздошних зчленувань, кістки таза без особливостей.
У клінічному аналізі сечі без патологічних змін, лише в окремих порціях помірна кількість оксалатів.
В уринолізісі зміни не виявлені.
Клінічний аналіз крові характеризувався в динаміці анемією від 104 г/л до 116 г/л і прискореної ШОЕ від 20 до 50 мм/год, інші показники не змінені.
У ТСХ сечі в динаміці високий рівень проліна - 52,36 мг/доба (норма 11,2-38,6), арнітин-аргінин-гістидинурія.
При високоефективній рідинній хроматографії визначалося зниження рівня гистідина і тріонина, інші амінокислоти - у межах нормальних показників.
Біохімічний профіль у динаміці характеризувався підвищенням рівня лужної фосфатази від 209,1 О/л до 240,7 О/л (при нормі до 104 О/л), підвищенням холестерину до 66,58 ммоль/л, АСТ від 41,4 до 60,2; зниженням рівня альбуміну до 32,19.
Дослідження серомукоїдів у динаміці - сіалові кислоти - 327-415-980 Од. (норма до 200), глікопротеїди - 0,74-0,79 (норма 0,25-0,45), кальцій - 2,3-3,0 ммоль/л (норма 2,2-3,2), лужна фосфатаза - 10,5-12,1-13,0 Од. (норма 2-5), холестерин 4,5 ммоль/л. (норма 3,6-6,2), тимолова проба 12,0 ОД (норма 0-4), β -ліпоптроеди 41 Ед. ( норма 35-45); глобуліни α 1-5,0% (норма 4-7), α 2 - 13,5% (норма 7-9), β- 16,2% (норма 9-14), γ -18,7% (норма 14-19).
Таблиця 2
Глікозаміно-глікани сироватки крові: |
11.02.02 |
10.06.02 |
11.06.02 |
13.11.02 |
26.06.03 |
23.04.04 |
Норма |
Загальні |
16,8 Од. |
4,7 мг./сутки (норма 3,5-5,5)
|
14,9 Од. |
16,0 Од. |
23,6 Од. |
29,8 Од. |
11,1-13,1 |
Розчинні сульфоглі-кани (хондраітін- 6-сульфати) |
10,6 Од. |
8,3 Од. |
8,4 Од. |
13,7 Од. |
20,0 Од. |
5,4-6,3 |
|
Середньо-розчинні сульфоглі-кани (хондраітін- 4-сульфати) |
5,0 Од. |
4,7 Од. |
5,7 Од. |
8,1 Од. |
7,9 Од. |
3,5-4,3 |
|
Трудно-розчинні сульфо-глікани |
1,2 Од. |
1,9 Од. |
1,9 Од. |
1,8 Од. |
1,9 Од. |
2,5-3,1 |
Система згортання крові - без змін.
У іммунограммі (30.01.2002): у клітинній ланці імунітету зниження Т-лімфоцитів зі збереженням реактогенності на іммунотропні препарати. Збільшення лімфоцитотоксичних аутоантитіл на тлі незначного зниження комплементарної активності, що свідчить про аутоіммуний компонент. Відзначається сенсибілізація до антигену вірусу герпес.
Таблиця 3
Імунограма хворої ДК
Показатели |
Результат |
Норма |
В лейкоцитів |
4,9х109/л |
4-8x109/л |
В нейтрофилів |
62%;3000 |
48-78%; 2000-5500 |
В лімфоцитів |
38%;1900 |
20-35%; 1200-3000 |
Т-лімфоцити (Е-РОК) СДЗ |
30%;570 |
40-75 %; 650-2000 68±2 |
Субпопуляції лімфоцитів СД4 СД8
|
24% 26% |
41±2 28±2 |
Співвідношення теофіллінчутливих і теофіллінрезистентних лімфоцитів |
1:0,92 |
1:2 |
В-лімфоцити (ЕАС-РОК) СД19 |
|
10-20% 10-15 |
Фагоцитоз: % активних клітин |
69% |
70-90% |
Мікробне число |
4,6 |
4,0-6,0 |
Імуноглобуліни IgА |
2,5 |
1,2-2,6 мг/мл |
IgG |
11,2 |
7,2-16,2 мг/мл |
IgM |
1,2 |
0,71-1,80 мг/мл |
IgЕ |
|
|
ЦИК: с 3,5% ПЕГ с 7%ПЭГ. |
0,05 |
не > 0,06 |
Лімфоцитотоксичні аутоантитіла |
24% |
до 10% |
Підбір: Реополіглюкин Неогемодез |
Р 13 Н 11 |
|
Комплемент |
0,9 |
1,0-1,1 |
Гемолізіни |
1,0 |
0,2-0,6 |
У імуннограмі (21.04.04.): Різке порушення співвідношення імуннорегуляторних клітин, яке приводе до дисбалансу (за рахунок зниження СД4 і підвищення СД8). Підвищення імунноглобулінів A і G на тлі високого ШОЕ (52 мм/ч) свідчить про активність запального процесу. Незважаючи на різке зниження хелперної ланки імунітету, що має, передусім, компенсаторний характер, на тлі аутоімунного процесу імунностимулююча терапія не показана.
Таблиця 4
Показники |
Результат |
Норма |
Загальна к-ть в лейкоцитів |
3,7х109/л |
4-8x109/л |
В нейтрофілів |
60%;2220 |
48-78%; 2000-5500 |
В лімфоцитів |
40%;1480 |
20-35%; 1200-3000 |
Т-лімфоцити (Е-РОК) СДз |
64%;947 |
40-75 %; 650-2000 68±2 |
Субпопуляції лімфоцитів СД4 СД8
|
19% 38% |
41±2 28±2 |
Співвідношення теофіллінчутливих теофіллінрезистентних лімфоцитів |
1:0,5 |
1:2 |
В-лімфоцити (ЕАС-РОК) СД19 |
17%
|
10-20% 10-15 |
Фагоцитоз: % активних клітин
|
84% |
70-90% |
Мікробне число
|
4,7 |
4,0-6,0 |
NK – клітини (CD16)
|
|
16-22 |
Імуноглобуліни IgА |
2,9 |
1,2-2,6 мг/мл |
IgG |
18,36 |
7,2-16,2 мг/мл |
IgM |
1,54 |
0,71-1,80 мг/мл |
IgЕ |
|
|
ЦИК: с 3,5% ПЭГ с 7% ПЭГ |
0,05 |
не > 0,06 |
Лімфоцитотоксичні аутоантитіла |
10% |
до 10% |
Підбір: Реополіглюкин Неогемодез |
|
|
Комплемент |
1,15 |
1,0-1,1 |
Гемолізіни |
0,95 |
0,2-0,6 |
При ЯМРТ шийного і грудного відділів хребта (13.06.2003р.) структурних змін у кістках не виявлено. Груба кіфотична деформація осі на рівні верхнього грудного відділу (імовірніше, за рахунок порушень у зв'язковому апараті) з помірно вираженими гіпотрофічними змінами в дисках, без дискогенної компресії хребетного каналу. За рахунок посилення кіфозу відзначається зниження висоти тіл хребців у передніх відділах. Спинний мозок не уражений, ліквороток не порушений.

Рис. 2.
Праве око: кришталик підвищеної ехогенності, деструктивні і фіброзні зміни склоподібного тіла, відслойка задньої гіалоїдної мембрани, у склоподібному тілі низько-ехогенна меко-дисперсна суспензія - запальний ексудат - увеїт, склоподібне тіло помутніле, зменшене, макула промінює до 1,6мм, розширений субретинальний простір з рідиною - киста в макулярній області.
Проведено молекулярну діагностику з використанням секвенування і ПДРФ-аналізу мтДНК, виділеної зі зразків крові і волосся. Виявлені поліморфізми 8697G/A, 8860G у гені тРНК-лізін та поліморфні гени MTHFR 677 C/T в гомозиготному стані. Результати молекулярної діагностики дозволили знайти і генотипову синтропію, яка реалізувалася у складну поєднану клінічну картину: моносомії Х, порушення обміну глікозаміногліканів та мітохондріальну дисфункцію на тлі дефіциту фолатного циклу.
На підставі проведеного дослідження встановлено діагноз: мозаїчна форма синдрому Шерешевського-Тернера в сполученні з мукополісахаридозом і вторинною мітохондріопатією, дефіцит ферментів фолатного циклу. Призначене лікування включало біоенергетичну терапію, ортопедичну реабілітацію, розроблена індивідуальна дієтотерапія, кофакторна терапія, санаторно-курортне лікування, масаж, кінезотерапія, що дозволили стабілізувати клінічну маніфестацію.
Зазначене спостереження є ілюстрацією нашого припущення про те, що мозаїчні форми хромосомних хвороб несуть у собі потенціал маніфестації того чи іншого метаболічного порушення, що і визначає подальший розвиток клінічних ознак. Важка травма, що мала місце, послужила ініціюючим фактором закладеного порушення обміну речовин, у даному спостереженні - глікозаміногліканів. Мітохондріопатія приєдналася на тлі важкої дезорганізації обміну сполучної тканини, а призначена біоенергетична терапія виявилася ефективною в комплексному симптоматичному лікуванні.
Та обставина, що у хворої були знайдені феномени порушення компактизації-декомпактизації та ознаки гіпометилювання, дала нам підставу запідозрити порушення функції регуляторних генів у хворої і висловити припущення при епігенетичний характер хвороби. Таке розуміння дозволило налагодити адекватну реабілітацію. Тригером у даному випадку була і герпес-вірусна інфекція.

Рис. 3. Хвора ДК до травми

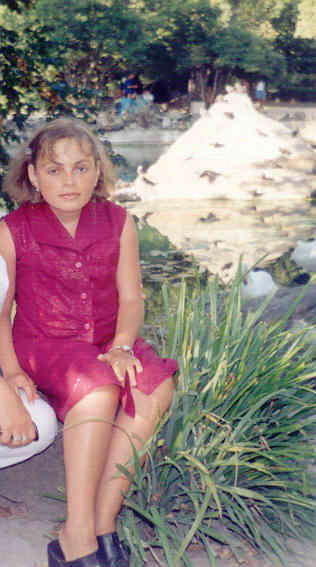
Рис. 4. Хвора ДК після першої травми

Рис. 5. Хвора ДК після другої травми
Хромосомні хвороби людини (ХХЛ) - одна з найдраматичних медичних і соціальних проблем, за якими стоять і репродуктивні втрати, і порушення психофізичного, соматичного й ендокринного стану, й психосоціальні конфлікти. ХХЛ відіграють зростаючу роль у патології людини на всіх етапах онтогенезу. Значна частина хромосомних хвороб відбирається природним добором на ранніх етапах внутрішньоутробного розвитку людини через спонтанні аборти, позаматкову вагітність. Існує обернено пропорційна залежність - хромосомні хвороби зустрічаються тим частіше, чим на більш ранньому етапі індивідуального розвитку ми їх шукаємо. До народження доходить лише частина хромосомних хвороб. Однак навіть ця частина є вражаючою по частоті - 1 з 150 народжених дітей має хромосомну аномалію.
Хромосомні хвороби (ХХ) (хромосомні синдроми) - це група вроджених спадкових захворювань, викликаних числовими або структурними абераціями хромосом, які видимі у світловому мікроскопі й характеризуються множинними вродженими вадами розвитку (МВВР).
Розвиток молекулярної цитогенетики відкрив можливості пошуку нових хромосомних синдромів, змінив уявлення про фенотипові особливості носіїв "малих аномалій хромосом" - хромосомного поліморфізму й хромосомної нестабільності.
Три мільярди пар підстав, що становлять геном людини, розподілені по 23 хромосомам. Кожна хромосома це гігантська молекула ДНК, з'єднана із численними спеціалізованими білковими молекулами. На кожній з 23 хромосом у лінійному порядку розташовані гени. Різні хромосоми відрізняються одна від одної кількістю пар нуклеотидів, кількістю ДНК і кількістю генів. Кількість пар нуклеотидів на хромосомах коливається від 50 до 250 мільйонів, у середньому - 150 млн.
Аномалії хромосом вивчає цитогенетика людини, а поєднання їх із клінічними проявами — клінічна цитогенетика. Хромосомні синдроми можуть бути зумовлені різними структурними аномаліями хромосом або зміною їхньої кількості. Числові зміни хромосомних наборів бувають у вигляді поліплоїдій (збільшення кількості хромосом, кратне гаплоїдному набору) або анеуплоїдій (збільшення або зменшення кількості хромосом у наборі, не кратне гаплоїдному). Порушення обох типів виникають внаслідок помилок мейозу і мітозу.
Цитогенетичні порушення за структурних перебудов трапляються у вигляді інсерції (вставки), делеції (втрати), дуплікації (подвоєння), інверсії (перевертання на 180°) і транслокації (переміщення) хромосом.
Інсерція — це вставка генетичного матеріалу внаслідок його переміщення.
Делеція — це структурна аномалія, що характеризується втратою термінальної або інтерстиціальної частини однією з гомологічних хромосом, — часткова моносомія у цій ділянці (рис. 6).
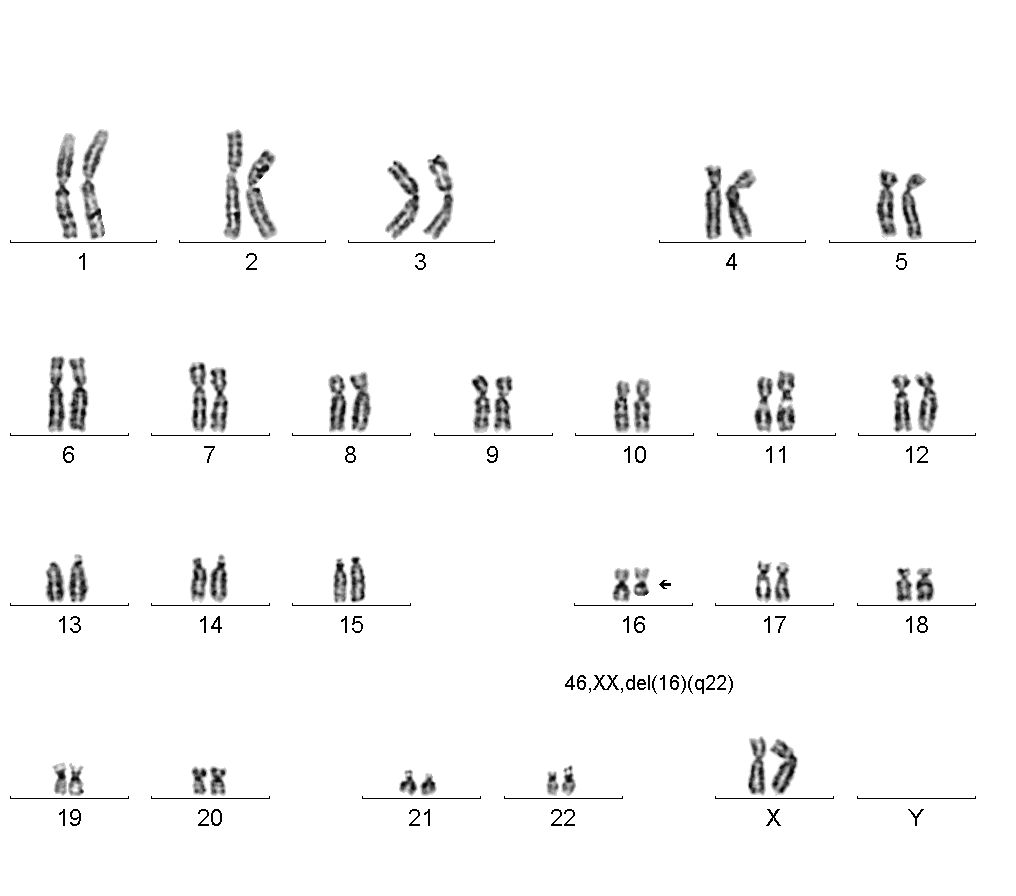
Рис.6. Часткова моносомія довгого плеча хромосоми 16
Транслокації хромосом виникають після розривів й обміну матеріалом між двома або кількома хромосомами. Вони бувають збалансованими і незбалансованими. Каріотипи, які мають увесь набір генів, але їхнє розташування в межах хромосом відрізняється від нормального, називають збалансованими хромосомними змінами. Збалансовані реципрокні аномалії хромосом можуть спричинити певні хромосомні синдроми. Описано випадки, коли деякі члени родини мали збалансований каріотип, тоді як інші — збалансовані реципрокні транслокації і виражену клінічну картину хромосомного синдрому. Прийнято вважати, що цей феномен пов'язаний з ефектом положення генів. Феномен ефекту положення генів полягає в тому, що гени, які опинилися поблизу точок розриву хромосом при формуванні збалансованої аномалії, змінюють свої прояви, тобто в одному оточенні генів їх функція збережена, а в іншому — порушена. У таких випадках є потреба в молекулярно-цитогенетичному дослідженні.
Збалансованою може бути реципрокна транслокація — взаємний обмін сегментами між двома негомологічними хромосомами без втрати залучених до нього хромосом (рис. 7). Збалансовані перебудови хромосом надзвичайно важливі в клінічній практиці, тому що нерідко діти — носії збалансованої хромосомної перебудови — фенотипово нормальні, але їхні нащадки мають високий ризик незбалансованих перебудов. В інших випадках транслокація між двома акроцентричними хромосомами з утратою ними коротких плечей призводить до утворення однієї метацентричної хромосоми замість двох акроцентричних. Такі транслокації називають робертсонівськими. Носії робертсонівських транслокацій мають моносомію по коротких плечах двох акроцентричних хромосом, але вони здорові, тому що втрата коротких плечей двох акроцентричних хромосом компенсується роботою таких же генів в інших 8 акроцентричних хромосомах. У носіїв робертсонівських транслокацій можуть утворюватися 6 типів гамет, з яких нулісомні гамети призведуть до моносомії за автосомами, а такі гамети не розвиваються. Носії збалансованого каріотипу самі можуть мати різні вади розвитку.
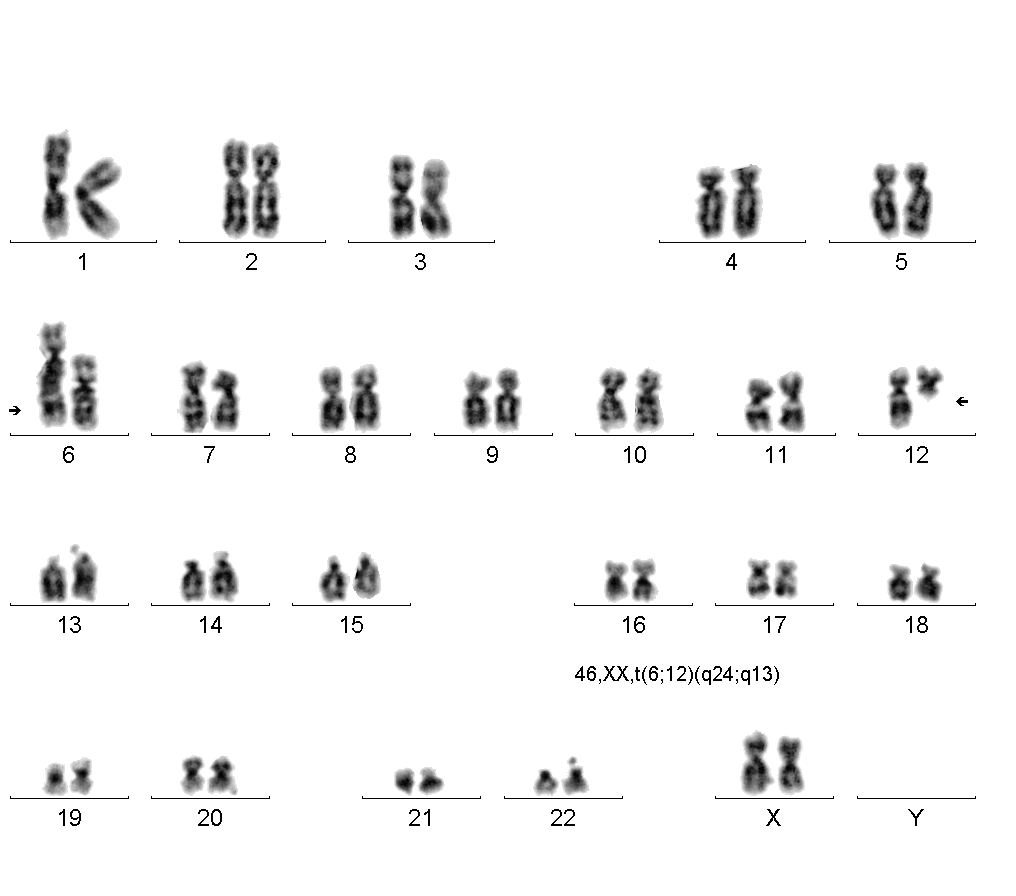
Рис. 7. Реципрокна транслокація

Рис.8. Робертсонівська транслокація
Кільцева хромосома — це структурна аномалія, що виникає внаслідок розриву довгого і короткого плечей хромосом, що призводить до втрати теломерних і злиття центромерних регіонів (рис.9).
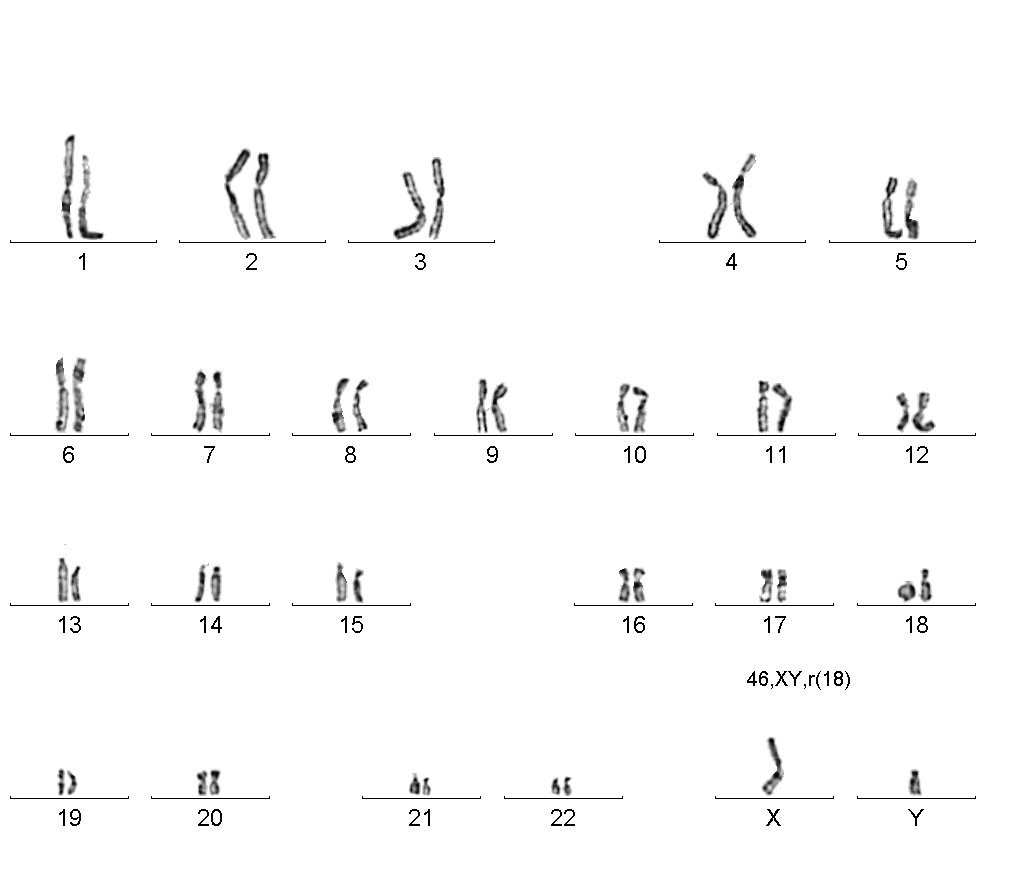
Рис.9. Кільцева хромосома
Дуплікація — це структурна аномалія, яка є наслідком подвоєння генетичного матеріалу, виникає в разі нерівного кросинговеру між сестринськими хроматидами і супроводжується патологічними фенотипами (рис.10).

Рис.10. Дуплікація
Дицентрична хромосома має 2 центромери, що утворюються внаслідок ізохроматидного розриву. При цьому теломерний акроцентричний регіон втрачається, а сестринські хроматиди з'єднуються. Після поздовжнього поділу центромери хромосома містить 2 центромери, дупліковану і делетовану частини (рис.11).

Рис.11. Дицентрична хромосома
Інверсія — це зміна лінійної послідовності генів на хромосомі внаслідок перевертання тієї або іншої її ділянки. Вони бувають перицентричними, коли до процесу залучається центромера, і парацентричними, коли перевертання відбувається поза зоною розташування центромери (рис.12).

Рис.12. Інверсія
Якщо хромосомна хвороба є результатом мутацій у гаметах батьків (що первинно виникли або успадковані ними від своїх батьків), ми говоримо про повну форму хромосомної хвороби, або гаметичні мутації. Якщо хромосомна хвороба виникає внаслідок мутації в клітинах ембріона на ранніх стадіях його розвитку, ми говоримо про неповні (мозаїчні) форми хромосомної хвороби, або зиготичні мутації. Мозаїчні форми можуть містити два, три і більше клонів з різним числовим співвідношенням.
Однією зі структурних хромосомних аномалій є ізохромосома, що утворюється внаслідок розриву хромосоми, яка проходить через центромеру (рис. 13).

Рис.13. Схема утворення ізохромосоми
Останніми роками чималий інтерес викликають мозаїчні форми хромосомних хвороб, спричинені неправильним поділом клітин на різних стадіях ембріонального розвитку. Мозаїчні форми хромосомних хвороб характеризуються легшим (порівняно з повною формою) перебігом, відсутністю окремих симптомів за наявності домінуючих ознак патології індивідуально залученої хромосоми. Легший перебіг мозаїчних форм пояснюється наявністю клонів нормальних клітин, які частково компенсують генний дисбаланс аберантних форм. Залежність тяжкості перебігу від співвідношення клонів точно не встановлена. Можливо, це пов'язано з тим, що не завжди вдається точно оцінити співвідношення клонів. Клінічні прояви мозаїчних форм варіабельні, що ускладнює встановлення діагнозу. Дослідження хромосом у лімфоцитах периферійної крові може бути неінформативним за незначної кількості аномальних клітин. Тому іноді для підтвердження діагнозу потрібно досліджувати хромосоми у фібробластах шкіри або в гонадах, використовуючи молекулярно-цитогенетичні методи.
Хромосоми людини зазнають впливу чинників навколишнього середовища і є надзвичайно чутливими до дії мутагенних чинників — радіації та хімічних речовин. У разі впливу таких чинників виникають порушення в соматичних або статевих клітинах. Через цитогенетичні порушення у соматичних клітинах страждає життєдіяльність усього організму. У разі впливу зазначених чинників на статеві клітини порушення хромосом проявляються в наступних поколіннях. Мутації хромосом у статевих клітинах призводять до утворення неповних аномальних гамет, у разі запліднення яких уможливлюється загибель зигот, ембріонів, народження дітей із хромосомними хворобами, для яких характерний певний фенотип. Мутації хромосом у соматичних клітинах призводять до неспецифічних хромосомних аномалій у вигляді хромосомних або хроматидних пробілів, розривів, обмінів у каріотипі, що не проявляються певним фенотипом, характерним для окремого хромосомного синдрому. Такі мутації не успадковуються. Вивчаючи вплив мутагенних чинників, можна якісно й кількісно оцінити дію йонізуючої радіації, хімічних речовин, вірусів, але отримані дані не можна переносити на статеві клітини, результатом дії на які є хромосомні хвороби. Вивчення дії мутагенних чинників нині є дуже актуальним з огляду на те, що кількість мутагенних чинників у навколишньому середовищі збільшується, а хромосомні соматичні мутації, що виникають у першому поколінні, нерідко спричинюють грубі порушення генного балансу в статевих клітинах і можуть становити серйозну небезпеку для спадковості майбутніх поколінь.
Формування клінічної картини хромосомної хвороби залежить від:
генотипу організму;
індивідуальності залученої в аберацію хромосоми або її ділянки (набір генів);
типу аберації;
розміру відсутнього (в разі делеції) або надлишкового (в разі часткової трисомії) матеріалу;
ступеня мозаїчності організму за аберантними клітинами;
умов середовища;
стадії онтогенезу або віку хворих.
Хромосомні хвороби мають загальні клінічні прояви:
черепно-лицеві дисморфії;
вроджені вади розвитку внутрішніх органів;
порушення росту й розвитку;
затримку психомоторного розвитку;
порушення функцій нервової та ендокринної систем.
Діагностичні ознаки хромосомних хвороб поділяють на три групи:
1-а група — комплекс ознак, які дають змогу запідозрити хромосомну аномалію. До них належать фізичне недорозвинення, деякі дисморфії мозку і лицьового черепа (деформація вушних раковин, епікант, високе піднебіння, мікроцефалія), клишоногість, клінодактилія мізинців, деякі вади внутрішніх органів;
2-а група — комплекс ознак, які виявляють у разі певних хромосомних захворювань: так, для трисомії 18 характерні доліхоцефалія, флексорне положення кистей, короткий і широкий палець (великий) стопи; для трисомії 13 — розколина верхньої губи і піднебіння, флексорне положення кистей, косоокість, дефект скальпа тощо;
3-я група — ознаки, характерні тільки для цієї хромосомної аномалії: котячий крик при синдромі 5р-, алопеція при синдромі 18р-.
Хромосомні хвороби характеризуються клінічним поліморфізмом, при цьому більш тяжка клінічна маніфестація спостерігається в разі аутосомних аберацій. Серед новонароджених дітей із хромосомними абераціями кількість тих, що мають аутосомні аберації, і тих, що мають аберації статевих хромосом, майже однакова, хоча автосом 22 пари, а статевих хромосом — одна пара. Це пов'язано з тим, що аутосомні аберації в переважній більшості випадків призводять до летального кінця ще до народження дитини.
Хромосомні хвороби спричиняють порушення генетичного балансу, і тому їхні патологічні ефекти виявляються на всіх етапах онтогенезу: проембріональному, ембріональному, фетальному і постнатальному у двох варіантах — летальному і вроджених вад розвитку. Летальний ефект хромосомної хвороби виявляється вже на стадії зіготи, він є однією з головних причин внутрішньоутробної загибелі плода. Установлено, що 30-40 % запліднених яйцеклітин гине на стадії бластули внаслідок різкого порушення морфогенетичних процесів. У процесі розвитку людини на ранніх стадіях бере участь приблизно 1000 генів, які локалізовані в усіх хромосомах. Хромосомна аномалія порушує взаємодію генів й інактивує певні процеси розвитку, такі як міжклітинні взаємодії, диференціювання клітин тощо.
Сумарний внесок хромосомних хвороб у внутрішньоутробну загибель плода становить 45 %. Що раніше переривається вагітність, то більша ймовірність хромосомної хвороби як причини порушення розвитку ембріона. Так, у 2-4-тижневого ембріона хромосомна хвороба виявляється у 60-70 %, в І триместрі вагітності — у 50 % абортусів, у II триместрі — у 25-30 %, у плодів, які загинули після 20 тиж. — у 7 %, у плодів, що загинули перинатально, — у 6%.
Отже, що менший термін вагітності, то частіше трапляються хромосомні аномалії. І що на більш ранньому періоді онтогенезу вони виявляються, то тяжчі форми дисбалансу хромосомного набору. Так, у ранніх абортусів, як правило, виявляють поліплоїдію (25 %), повні трисомії за автосомами (50 %).
Автосомні трисомії 1, 5, 6, 11 і 19 хромосом припиняють розвиток плода в доімплантаційний період, порушують гаметогенез і тому виявляються рідко.
Моносомії за автосомами призводять до загибелі на стадії гамет і зигот.
Хворі зі структурними хромосомними хворобами здебільшого виживають.
Хромосомні аномалії спричинюють також ефекти, які простежуються протягом усього життя. Так, хромосомні хвороби в соматичних клітинах можуть бути для них нейтральними, зумовлювати їхню загибель, активізувати поділ клітин, змінювати їхню функцію.
Хромосомні хвороби соматичних клітин трапляються в 1—3 % випадків, здебільшого вони елімінуються імунною системою. Але в деяких випадках вони можуть стати причиною злоякісного росту внаслідок активізації онкогенів за наявності транслокацій, делецій. Так, транслокації між 9 і 22 хромосомами спричинюють мієлолейкоз. Опромінення і хімічні мутагени збільшують кількість хромосомних аберацій у соматичних клітинах.
Хромосомні аберації накопичуються також у процесі старіння.
Патогенез хромосомних хвороб донині не з'ясований. Однак за будь-яких трисомій і часткових моносомій можна виділити три типи генетичних ефектів:
специфічні;
напівспецифічні;
неспецифічні.
Специфічні ефекти пов'язані зі зміною кількості структурних генів, які кодують синтез білка. Так, у разі трисомії 21 у 50% хворих виявлено підвищення активності надпероксиддисмутази — гена, локалізованого на хромосомі 21.
Подібний "ефект дози гена" виявлено для кількох десятків генів за трисомій на різних хромосомах. Однак біохімічна зміна фенотипу хромосомної хвороби поки що не дає змоги зрозуміти шляхи патогенезу вроджених порушень, які виникають унаслідок хромосомних аномалій. Виявлені біохімічні відхилення поки що важко порівнювати з фенотипними характеристиками хвороб на органному й системному рівнях. Зміна кількості алелів гена не завжди спричиняє пропорційну зміну продукування відповідного білка. У разі хромосомних хвороб завжди істотно змінюється активність інших ферментів або кількість білків, гени яких локалізовані на не залученій у дисбаланс хромосомі. У жодного пацієнта з хромосомною хворобою не виявлено білка-маркера.
Напівспецифічні ефекти зумовлені зміною кількості генів і в нормі представлені численними копіями; до таких генів відносять рибосомні і транспортні РНК, гістонові і рибосомні білки, скоротливі білки актину й тубуліну. Ці білки в нормі контролюють ключові етапи метаболізму клітини, її поділу, міжклітинних взаємодій. Які фенотипні ефекти дисбалансу цієї групи генів, як компенсується їх дефіцит або надлишок, досі залишається невідомим.
Неспецифічні ефекти хромосомних аномалій пов'язують зі зміненим вмістом гетерохроматину в клітині. Важлива роль гетерохроматину в клітинному поділі, рості та інших біологічних функціях не викликає сумнівів.
Таким чином, неспецифічні і, частково, напівспецифічні ефекти наближають нас до розуміння клітинних механізмів патогенезу.
Патогенез хромосомних хвороб розгортається в ранній внутрішньоутробний і триває в постнатальний період. Множинні вади розвитку як головні фенотипні прояви хромосомних хвороб формуються в ранньому ембріогенезі, тому до періоду постнатального онтогенезу спостерігалися всі основні вади розвитку.
Жодних специфічних рис патогенезу хромосомних хвороб не виявлено ані на молекулярному, ані на клітинному рівні. Характерна риса хромосомного дисбалансу — множинність вад розвитку, що уражують різні органи й системи. За кожної форми хромосомної хвороби спостерігається 30—80 різних відхилень від норми. Деякі хромосомні хвороби характеризуються лише певним поєднанням відхилень у розвитку, а не специфічними вадами.
У разі хромосомних хвороб відхилення від нормального розвитку корелюють зі ступенем хромосомного дисбалансу. Чим більше хромосомного матеріалу залучено до мутації, тим раніше захворювання виявиться в онтогенезі, і тим значнішими будуть порушення у фізичному і психічному розвитку індивіда. Як правило, надлишок хромосом (або частин їх) є набагато сприятливішим, ніж дефіцит. У міру вивчення фенотипів і різних хромосомних мутацій з'ясовується, що найспецифічніші для того або іншого синдрому особливості зумовлені порівняно незначними відхиленнями у вмісті хромосомного сегмента. Дисбаланс у значному обсязі хромосомного матеріалу дає більш неспецифічну клінічну картину. Так, специфічні клінічні симптоми синдрому Дауна спостерігаються в разі трисомії за сегментом довгого плеча хромосоми 2q22.1. У разі синдрому котячого крику при делеції короткого плеча хромосоми 5 за розвиток клінічної картини відповідає сегмент 5р15. Характерні риси синдрому Едвардса пов'язані з трисомією ділянки хромосоми 18q11.
Хромосомні аномалії спричинюють порушення загального генетичного балансу зкоординованості в роботі генів і системної регуляції, які склалися в процесі еволюції для кожного виду.
Специфічність патогенезу багатьох спадкових і неспадкових хвороб визначається станом імунної та ендокринної систем організму, функції яких генетично детерміновані. Несприятливий спадковий фон може бути провокаційним моментом у розвитку будь-якої патології.
Приблизна частота хромосомних хвороб становить 0,59 %: анеуплоїдії, включаючи мозаїчні форми, трапляються в 38 % хворих із хромосомними порушеннями; трисомії за аутосомами — у 22 %; структурні перебудови — у 40 % (1/2 структурних перебудов — сімейні випадки).
Усі трисомії - спорадичні випадки і є результатом мутацій де ново.
Останнім часом з'явилися принципово нові підходи до діагностики хромосомної патології. Це стало можливим завдяки розробленню і впровадженню в клінічну цитогенетику нових технологій, таких як ДНК-діагностика, гібридизація нуклеїнових кислот іn situ і комп'ютерні системи для аналізу хромосом. ДНК-діагностика грунтується на використанні технології рекомбінантних молекул ДНК і приготуванні спеціальних проб для виявлення і молекулярного аналізу генетичних дефектів. Флуоресцентна гібридизація іn situ (FISH) включає застосування спеціально підготовлених ДНК-проб для виявлення генетичних дефектів на хромосомному рівні і проведення процедури молекулярно-цитогенетичної діагностики з використанням сучасної флуоресцентної мікроскопії.
Комп'ютерна система для аналізу хромосом передбачає наявність спеціальних телекамер для виявлення надслабких сигналів, отриманих при проведенні гібридизації іn situ, а також програм, які дають змогу проводити автоматичний аналіз хромосом і високоефективну кольорову детекцію ДНК-зондів для діагностики хромосомних і генних порушень на мікроскопічному рівні.
Ідентифікація хромосомної патології ґрунтується на використанні ДНК-зондів різних типів, які дають змогу маркувати індивідуальні хромосоми або окремі ділянки хромосом. ДНК-зонди, як правило, - клоновані фрагменти геному людини або їх окремі ділянки. Шляхом гібридизації ДНК-зондів з метафазними хромосомами або інтерфазними клітинами визначають особливості хромосомного набору і наявність навіть незначних хромосомних порушень, які не можна виявити за допомогою класичних цитогенетичних методів діагностики.
Патологія у системі аутосом перебігає тяжче, ніж аномалії за статевими хромосомами. Це, ймовірно, пов'язано з різною генетичною активністю хромосом: У-хромосома несе мало генів; одна Х-хромосома в жінки перебуває в неактивному стані.
Величезну роль у діагностиці хромосомних хвороб відіграють соматогенетичні дослідження із синдромологічним аналізом, які розроблені патологоанатомом Е.Д. Черствим. Цей універсальний метод, що його започаткував Моргані, дав змогу скласти чітку систему опису фенотипу в постнатальний період онтогенезу. Переконаність у високій точності цього методу стала передумовою застосування його для пренатальної оцінки дизморфогенезу за допомогою ехографії.
Класифікація хромосомних хвороб відповідно до залученої хромосоми постійно поповнюється через безупинне збільшення кількості описаних хромосомних синдромів.
Причиною хромосомних хвороб є хромосомні і геномні мутації (триплоїдія, тетраплоїдія, анеуплоїція). Із геномних мутацій у людини трапляються тільки трисомії за аутосомами, полісомії за статевими хромосомами (М.П. Бочков, 2001). Хромосомні мутації характеризуються значною різноманітністю.
Нозологічна форма хромосомної патології повинна визначатися тільки на основі етіології — характеру генетичної події (нестачі або надлишку хромосомного матеріалу) та її обсягу (залучення до патологічного процесу хромосоми, сегмента).
Секвенування генома дозволило встановити локалізацію генів, асоційованих з різними захворюваннями на карті хромосом.
Нижче наведена характеристика фенотипів хворих на хромосомну патологію у відповідності із класифікацією хромосом. В рисунках хромосом визначена локалізація патологічних генів, які пов’язані із різними спадковими хворобами.
М.Рідлі (2008) охарактеризував 23 хромосоми з позицій не тільки побудови, а й функції окремих районів кожної із них, як найбільш доведених і значущих у житті людини. Саме тому при розгляді клінічних особливостей хромосомних хвороб, ми використали його загальні характеристики кожної хромосоми.