В ответ на уменьшение МОС рефлекторно увеличивается синтез альдостерона повышается реабсорбция Na повышается реабсорбция воды увеличивается ОЦК
Билет 60
2. Гипероксия. Причины возникновения, формы. Механизмы повреждающего действия кислорода. Роль свободно-радикальных процессов. Понятие о системе антиокислительной защиты организма.
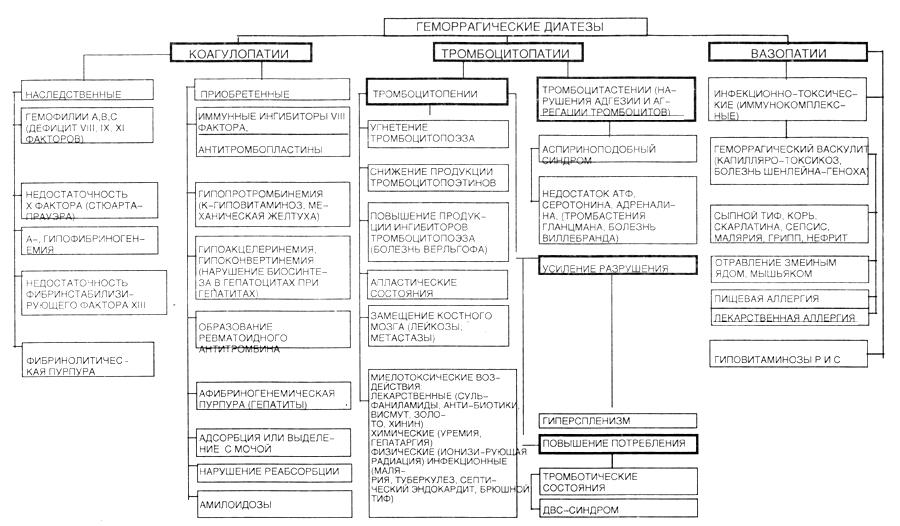
3. Виды, этиология, патогенез и клинические проявления геморрагических диатезов. Роль наследственных факторов, а также иммунопатологических механизмов в их патогенезе.
Геморрагические диатезы.
Разделяются в зависимости от повреждения компонента гемостаза:
Вазопатии:
Наследственные:
телеангиэктазия или болезнь Рандю-Ослера – наследуется аутосомно-доминантно, т.е дефект структуры сосудистой стенки, она истончается и образуются мешкообразные эктазы. Локализация: губы, верхняя часть туловища, лицо, волосистая часть головы, на слизистой оболочке, в полостях. Эти истонченные стенки хорошо травмируются. У пациента формируется хроническая анемия железодефицитная вследствие хронической кровопотери.
Приобретенные:
Гиповитаминозы С (цинга), РР – необходимы для полимеризации стенки сосудов – у пациента образуются петехии, васкулит. На симметричных участках кожи.
Геморрагичсекий васкулит (болезнь Шенлейна-Геноха):
Причина: бактерии, вирусы, паразиты, вакцины, лекарственные препараты, пищевые продукты. Заболевание имеет иммунокомплексный механизм, формируются циркулирующие иммунные комплексы – 3 тип аллергии. Имеет сезонный характер – весна. ЦИК – очень крупные, повреждают эндотелий сосудов, и способствуют образования воспалении. Результат – развитие иммунного воспаления. Формы: кожная, суставная. Если присутствуют на брюшине – абдоминальная форма, у пациента боли в животе. Эти ЦИК любят клубочки почек – почечная форма, формируется почечный синдром с симптомами гломерулонефрита.
Пробы на резистентность сосудистой стенки: Кончаловского (до определенного значения нагнетается давления в манжете, затем подсчитывается количество петехий), жгута (после наложения манжеты при измерении АД, смотрят на кожу – будут кровоизлияния), щипка (ущипнуть кожу).
При геморрагических вазопатиях поражение сосудистой стенки,приводящие к нарушению тромбоцитарно-сосудистого гемостаза и кровоточивости, возникает вследствие повышения проницаемости стенки кровеносных сосудов и ее деструкции.
Тромбоцитопатии:
Тромбоцитопении:
Болезнь Верльгофа (тромбоцитопеническая пурпура) – наследуется аутосомно-доминантно. Проявляется в виде синяков, причем цветут – кожа леопарда.
По наследству передается способность селезенки вырабатывать ингибитор тромбоцитопоэза (селезеночный фактор). Этот фактор ингибирует отшнуровку тромбоцитов от мегакариоцитов в костном мозге, количество мегакариоцито возрастает, а толку от них мало.
Лечат: удалением селезенки.
Приобретенные: НА ТЕЛЕФОНЕ ФОТО!
Миелотоксическое воздействие:
Экзогенные факторы: физические (ионизирующая радиация), химические (цитостатики, антибиотики, продукты нефтепереработки), биологические (вирусы, токсины микроорганизмов).
Эндогенные:
Угнетение нормальных ростков кроветворения.
Аутоиммунная тромбоцитопения.
Результат – носовые, десневые кровотечения, маточные, синяки.
Для дифференцировки существует биопроба, животному вводится сыворотка больного. Селезеночный фактор не обладает видоспецифичностью, поэтому он вызывает тромбоцитопению и у животного.
Тромбоцитастении – нарушение функциональных свойств тромбоцитов:
Наследственные: болезнь Гланцмана, болезнь Виллебранда (комплексный геморрагический диатез, потому что эндотелий в норме вырабатывает фактор Виллебранда, составную часть 8 фактора,. А по наследству передается извращенный синтез этого фактора. Фактор Виллебранада – фактор коагуляции).
Приобретенные:
Гиповитаминоз С, В12.
Уремия.
Переливание больших доз крови, плазмы, концентратов прокоагулянтов.
Миеломная болезнь, болезнь Вальденстрема (повышение в поазме нормальных и аномальных белков).
ДВСС (повышение ПДФ – продукты деградации фибрина)
Лекарственные - НПВС
Механизм тромбоцитоастений:
Нарушение синтеза и накопление в гранулоцитах тромбоцитов БАВ,
Нарушение дегрануляции и высвобождения тромбоцитарных факторов в плазм крови.
Диагностика тромбоцитопений и астений:
Количество тромбоцитов: 180-400*109/л.
Время агрегакции тромбоцитов: 14-18 сек
Процент клеток, вступивших в агрегацию – более 90.
Удлинение времени и уменьшение клеток вступивших – тромбоциастения.
Положительная проба Кончаловского – тромбоциты определяют трофику эндотелия сосудов, выделяют факторы роста необходимы для эндотелиоцитов. Если тромбоцитов мало – дефекты, ломкость сосудов повышается.
Патогенез: тромбоцитопения-уменьшение сод-я тромбоцитов в крови ниже нормы. Тромбоцитоастения- качественная неполноценность и дисфункция тромбоцитов при нормальном или пониженном их содержание.
Выделяют 4 основных механизма возникновения тромбоцитопений: умен. продукции,усиленное разрушение, повышенное потребление,перераспределение тромбоцитов. Нарушение гемостаза и развитие кровоточивости при тромбоцитопении обусловлено следующими мех-ми: 1) повыш. Проницаемости микрососудов для эритроцитов и др. составных частей крови и ломкости сосудов вследствие дистрофии стенки при выключение ангиотрофической ф-ции тромбоцитов.
2)умен. адгезивно-агрегационной ф-ции тромбоцитов.
3) нар. Р-ции освобождения тромбоцитарных факторов свертывания крови,АДФ, серотонина, адреналина, антигепаринового фактора,следствием чего является
Коагулопатии – нарушение коагуляционного гемостаза:
Наследственные. По фазам свертывания:
Нарушение I фазы свертывания:
Гемофилия А (80% сех гемофилий) – дефицит VIII фактора.
Гемофилия В (10%) – дефицит IX фактора.
Гемофилия С (5%) – дефицит XI фактора.
Механизм наследования гемофилии: гемофилия А – это дефект X хромосомы. Здоровый отец, мама – носитель: здоровый сын, здоровая дочь, дочь-носитель, страдающий гемофилией сын. Заболевание проявляется сразу после рождения, у новорожденного возможны различные кровоизлияния (кефалогематома). Любая травма приводит к образованию гематом (тип кровоизлияния). Возможно формирование гемартрозов.
В и С гемофилии могут быть и у мужчин и у женщин.
Диагностика нарушений I фазы свертывания:
Активированное время рекальцификации плазмы (АВР) 40-60сек.
Активированное частично тромбопластиновое время (АЧТВ) 33-45сек.
Нарушение II фазы:
Наследственные (парагемофилии): гипопротромбинемия, гипопроконвертинемия, гипопроакцелеринемия.
Приобретенные: печеночная недостаточность, ахолия, гиповитаминоз К.
Чтобы оценить состояние II фазы оцениваем протромбиновый индекс (ПТИ) 85-110%.
Нарушение III фазы:
Наследственные: гипофибриногенемия, афибриногенемия, дисфибриногенемия.
Приобретенные в результате либо в уменьшении синтеза, или повышенное потребление (особенно при ДВСС): гипофибриногенемия, афибриногенемия.
Оценка III фазы:
Тромбиновое время (в плазму пациента добавляют тромбин): 11-18сек.
Фибриноген: 2-4 г/л.
Все геморрагические диатезы разделяют от того, на какой фазе свертывания происходит нарушение:
IV фаза – фибринолиз – активация может послужить основой диатеза (онечный гиперфибринолиз):
Наследственные – формирование фибринолитической пурпуры.
Приобретенные – лечение фибринолитиками – повышение тканевых активаторов плазминогена (опухоли, стресс, почечная, печеночная недостаточность).
Оценить состояние IV фазы можно:
Спонтанный эуглобулиновый лизис - 2-4 часа.
Продукты деградации фибрина (ПДФ) 4-6 мг/л
Тромбоэластограмма: если активность фибринолитической системы повышена – время сокращается (спонтанный глобулиновый индекс), и возрастает количесвто продуктов деградации.