

3.3 Влияние температуры на скорость реакции. Энергия активации
В общем случае
скорость химической реакции с повышением
температуры увеличивается. Поскольку
![]() ,
то влияние температуры на скорость
выражается через влияние температуры
наk,
поскольку концентрация от температуры
практически не меняется. Опыт показывает,
что при повышении температуры на 10 К
скорость реакции возрастает в 2…4 раза
(правило Вант-Гоффа). Для характеристики
зависимости скорости химической реакции
от температуры был выведен температурный
коэффициент
скорости реакции:
,
то влияние температуры на скорость
выражается через влияние температуры
наk,
поскольку концентрация от температуры
практически не меняется. Опыт показывает,
что при повышении температуры на 10 К
скорость реакции возрастает в 2…4 раза
(правило Вант-Гоффа). Для характеристики
зависимости скорости химической реакции
от температуры был выведен температурный
коэффициент
скорости реакции:
![]() .
.
Если известны константы скорости при температурах, отличающихся не на 10 К, то коэффициент Вант-Гоффа может быть определен из уравнения:
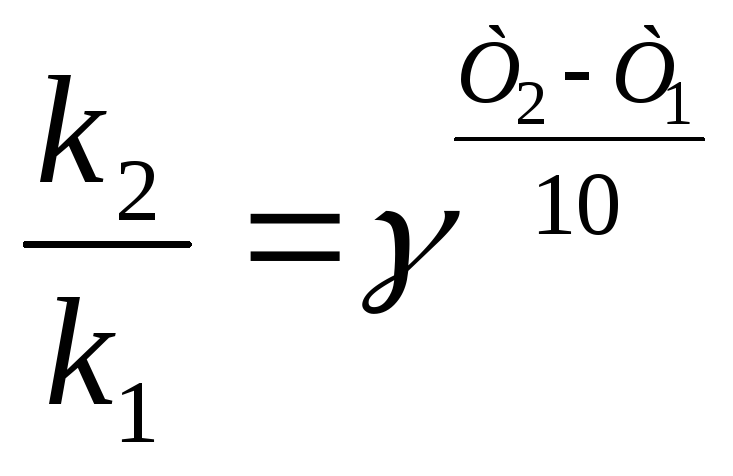 ,
,
где k2 и k1 – константы скорости при температурах Т2 и Т1.
Более строгую зависимость константы скорости от температуры дает уравнение:
![]() ,
(3.6)
,
(3.6)
которое получило название уравнения Аррениуса. Величина Е имеет размерность энергии и носит название энергии активации.
Энергию активации
можно определить как тот избыток
энергии по сравнению со средней энергией
молекул при данной температуре, которым
должны обладать молекулы, чтобы они
могли легко вступить в химическую
реакцию. Поскольку Е > 0, то производная
![]() >0
следовательно,lnk
и k
будут всегда возрастать
с ростом Т.
>0
следовательно,lnk
и k
будут всегда возрастать
с ростом Т.
Связь энергии
активации с тепловым эффектом можно
проиллюстрировать с помощью представления
об энергетическом барьере (рисунок
3.6). Химическую реакцию можно представить
как переход системы из энергетического
состояния 1 в состояние 2, сопровождающееся
тепловым эффектом
![]() (при Р =const).
Из рисунка 3.6 видно, что переход из
состояния 1 в состояние 2 возможен при
затрате энергии Е, а обратный переход
возможен при затрате энергии Е´.
Поскольку Е´
> Е, то энергетический барьер обратной
реакции будет больше, чем прямой.
Следовательно скорость обратной реакции
будет меньше скорости прямой.
(при Р =const).
Из рисунка 3.6 видно, что переход из
состояния 1 в состояние 2 возможен при
затрате энергии Е, а обратный переход
возможен при затрате энергии Е´.
Поскольку Е´
> Е, то энергетический барьер обратной
реакции будет больше, чем прямой.
Следовательно скорость обратной реакции
будет меньше скорости прямой.
В задачу химической кинетики входит поиск способов уменьшения энергетического барьера, энергии активации, для увеличения скорости. Одним из таких способов является применение катализатора.
При осуществлении
реакции в прямом направлении выделяется
количество энергии
![]() ,
а в обратном направлении затрачивается
такое же количество энергии, что
выделяется в прямом.
,
а в обратном направлении затрачивается
такое же количество энергии, что
выделяется в прямом.
Уравнение (3.6) легко
проинтегрировать. Считая, что
![]() ,
получим
,
получим
![]() ,
(3.7)
,
(3.7)
где lnA – константа интегрирования.
![]() .
.
Отсюда видно, что величинами, характеризующими реакцию, являются предэкспоненциальный множитель А и энергия активации Е.
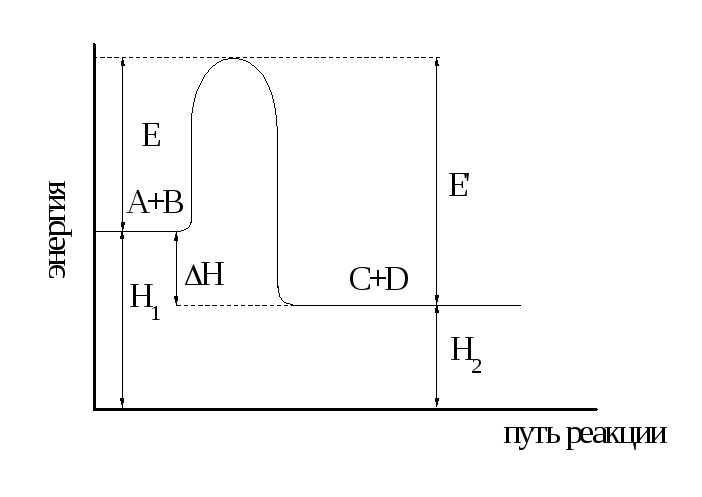
Рисунок 3.6 –
Диаграмма, иллюстрирующая связь Е
и
![]()
Поскольку для вступления в химическую реакцию молекулы должны обладать некоторой избыточной энергией (Е) по сравнению со средней величиной (рисунок 3.7), то влияние повышения температуры на рост скорости реакции можно объяснить увеличением числа частиц, обладающих этим избытком энергии.
Обратимся к рисунку
3.7, где изображена зависимость величины
![]() от энергии, так называемая кривая
распределения молекул по энергиям. ГдеdN
– число молекул, обладающих энергиями
от
от энергии, так называемая кривая
распределения молекул по энергиям. ГдеdN
– число молекул, обладающих энергиями
от
![]() до
до![]() ;N0
– общее число молекул. Пусть
;N0
– общее число молекул. Пусть
![]() - энергия, превышающая среднюю на энергию
активации.
- энергия, превышающая среднюю на энергию
активации.
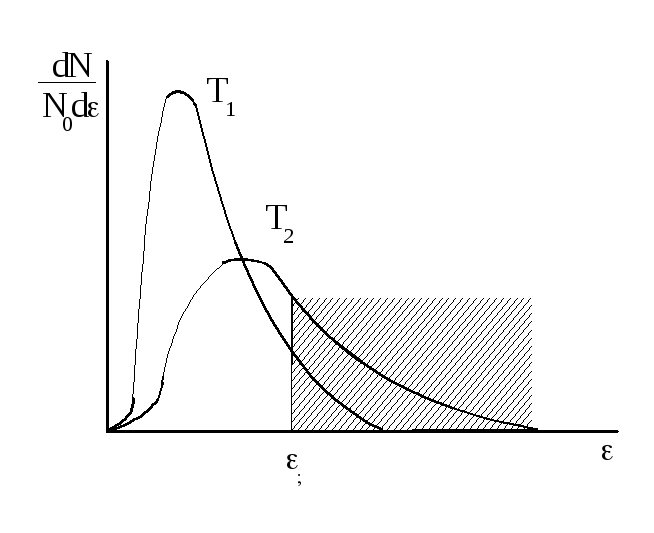
Рисунок 3.7 – Кривая распределения молекул по энергиям
Площадь под кривой
правее абсциссы
![]() ,
ограниченная кривой и осью абсцисс,
определяет собой долю молекул, обладающих
энергией, превышающей
,
ограниченная кривой и осью абсцисс,
определяет собой долю молекул, обладающих
энергией, превышающей![]() .
С повышением температуры (Т2
>Т1)
кривая сдвигается вправо, и эта доля
молекул быстро возрастает.
.
С повышением температуры (Т2
>Т1)
кривая сдвигается вправо, и эта доля
молекул быстро возрастает.
Величину энергии активации можно определить двумя методами. Первый метод – графический, с использованием уравнения Аррениуса в виде (3.7):
![]() .
.
Если построить
график зависимости экспериментальных
величин ln
k
от 1/Т, то получим прямую. По оси ординат
отсекается отрезок, равный lnA.
Тангенс угла наклона на кривой равен
![]()
![]()
![]()
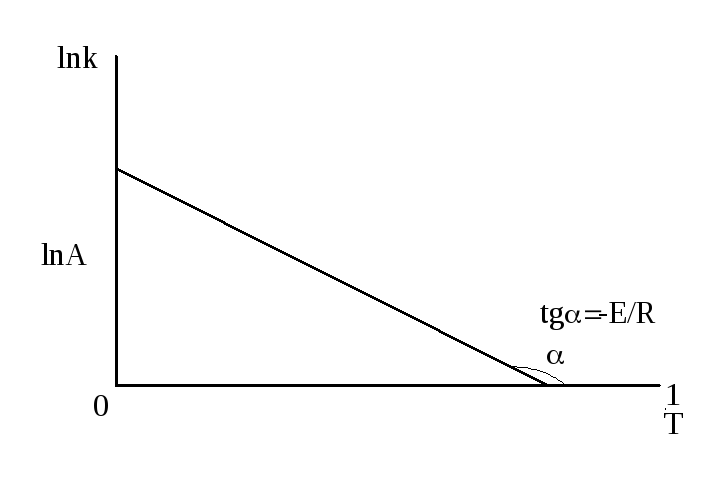
Рисунок 3.8 – Зависимость логарифма константы скорости химической реакции от обратной температуры
Второй метод основан на измерении скорости химической реакции при двух температурах. Для этого из уравнения (3.6) получим:

Постоянной энергия
активации может быть только в простых
реакциях. Для сложных реакций величина
Е
является переменной и не имеет такого
простого физического смысла, как в
случае простых реакций. Тем не менее, и
в этом случае, принято величину Е
называть энергией активации, но определяют
ее из дифференциального уравнения
Аррениуса
![]() ,
используя зависимость
,
используя зависимость![]() и
и![]() .
.
Для сложных реакций,
следовательно, не будет линейной
зависимости
![]() .
.