

1.2.1. Уравнение состояния термодинамической системы. Нулевой
закон термодинамики
Свойства, определяющие состояние системы, взаимосвязаны между собой. Уравнение, связывающее термодинамические параметры системы в равновесном состоянии функциональной зависимостью называется уравнением состояния (УС). Например, для однородного чистого вещества в отсутствии электрических, магнитных и иных полей и когда можно пренебречь поверхностной энергией, такое уравнение имеет общий вид P=f(V,T). Это уравнение называют термическим УС. Уравнение вида U=f(V,T) (U–внутренняя энергия) называют калорическим УС. Для полного термодинамического описания системы обязательно надо знать её УС.
Существование УС (а также самого понятия «температура») вытекает из закона термического равновесия (нулевого закона термодинамики):
Две системы, находящиеся в термическом равновесии (т.е. когда в отсутствии теплоизоляции не происходит теплообмена между системами) с третьей системой, состоят в термическом равновесии друг с другом.
Этот закон был назван «нулевым» из-за того, что он был сформулирован Р. Фаулером только в 1931 году, т.е. значительно позже открытия первого и второго законов термодинамики.
1.2.2. Идеальный газ и его уравнение состояния
Физическая химия широко использует для описания систем и процессов модельные представления. Модель обязательно должна учитывать основные свойства моделируемого явления или объекта. Так, важнейшая термодинамическая система физической химии идеальный газ – модель, описывающая разряженный реальный газ, т.е. газ, находящийся при достаточно низком давлении и относительно высокой температуре. При этих условиях можно пренебречь силами притяжения и отталкивания между частицами газа, а также их собственными размерами по сравнению со средними межчастичными расстояниями. Частицы идеального газа (материальные точки) сталкиваются между собой упруго, обмениваясь при этом кинетической энергией.
Простейшим УС, имеющим огромное значение для всей физической химии, является известное Вам уравнение состояния идеального газа или уравнение Менделеева-Клапейрона:
P=nRT/V, (2.1)
или Z=PVm/RT=1, (2.2)
где n – количество вещества газа, моль; R – универсальная газовая постоянная, равная 8,314 (Дж/моль. К); Z - коэффициент сжимаемости.
1.2.3. Реальный газ и его уравнения состояния
Опыт показывает, что с увеличением давления и понижением температуры поведение реального газа всё в большей степени отклоняется от поведения идеального газа. Эти экспериментальные факты объясняются тем, что молекулы реального газа взаимодействуют между собой и имеют собственный объём.
Взаимодействие между частицами в основном включает электростатические силы притяжения (межмолекулярные силы или, иначе, силы Ван-дер-Ваальса) и силы отталкивания между частицами. Наличие сил Ван-дер-Ваальса равноценно дополнительному сжатию газа, т.е. добавочному давлению, которое следует прибавить к внешнему давлению Pвн. Это добавочное давление называется внутренним или статическим давлением Pст=n2a/V2, где a – параметр, характеризующий межмолекулярные силы. Следовательно, общее давление можно выразить уравнением:
P=Pвн + Pст. = Pвн + n2a/V2 . (2.3)
Для учёта собственного объёма частиц примем, что частица реального газа ограничена некоторой поверхностью, непроницаемой для других частиц. Это значит, что реальный газ нельзя сжать до бесконечности. Следовательно, для газа доступен не весь объём сосуда V, а только часть его – свободный объём V*, равный
V*=V-nb, (2.4)
где b – запрещённый объём в расчёте на 1 моль газа.
Простейшее из уравнений состояний реального газа – уравнения Ван-дер-Ваальса – получается модификацией уравнения Клапейрона-Менделеева - давление Р и объём V заменяются выражениями (2.3,4):
(Pвн+ n2a/V2)(V-nb)=nRT (2.5)
На рис.1 показаны реальные изотермы на примере углекислого газа.
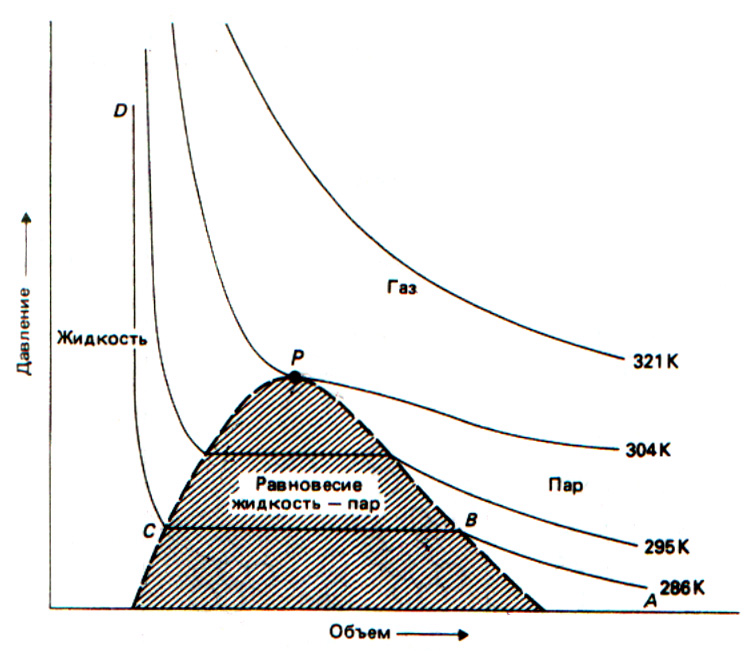
Рис.1.1. Изотермы углекислого газа
На участке ABпри повышении давления происходит сжатие газа, в точкеBнаступает его конденсация в жидкость, и газ правильнее назватьнасыщенным паром. Вместо участкабвгд (рис. 2.6) на реальной изотерме имеется горизонтальный участокBC, соответствующий двухфазной системе «пар-жидкость». На участкеBCпроисходит конденсация пара и уменьшение объёма при постоянном давлении. От точкиCдавление резко увеличивается, что связано с малой сжимаемостью жидкости.
С ростом температуры двухфазная область становится всё уже. При некоторой температуре участок ВС вырождается в точку (критическая точка). Этой точке отвечаеткритическое состояние, которое характеризуетсякритическими:температуройTk , давлениемPkи мольным объёмомVk. В области критического состояния возрастает вероятность возникновения различных микроскопических сгущений вещества –флуктуацийплотности. Эти флуктуации приводят к образованию в паре микроскопических капелек жидкости, а в жидкости – микроскопических пузырьков пара. Флуктуации плотности проявляются в опалесценции – сильном рассеянии света на капельках и пузырьках. При температуре выше критической газ нельзя обратить в жидкость никаким сжатием. Значения критических параметров для некоторых веществ приведены в табл.3 П4 (раздел 4).
Данные о критических параметрах позволяют оценить: значения параметров а и b в уравнении Ван-дер-Ваальса. Метод расчёта вытекает из анализа уравнения Ван-дер-Ваальса (для 1 моль газа), приведённого к кубическому виду:
V3 – (b+RT/P)V2 + (a/P)V –ab/P=0 (2.5)