105. Врожденный гипотериоз. Лечение
Лечение Заместительная терапия показана и эффективна при любой форме гипотиреоза кроме крайне редких случаев нечувствительности тканей к тироксину. Лечение начинают с момента установления диагноза независимо от возможности лабораторного подтверждения. Допускается лечение с периода новорожд. до 1-2-летнего возраста с последующей отменой препаратов и обследованием. Первоочередная цель при лечении ВГТ — как можно быстрее повысить уровень Т4до нормы, чтобы предотвратить или свести к минимуму поражение ЦНС. Обычно используют левотироксин в стартовой дозе 10-14 мкг/кг/сут. Считают, что такая доза безопасна и не вызывает гипертиреоз у новорожденных и детей раннего возраста. При этом показано, что уровень Т4 нормализуется уже через 1 нед. Однако в ряде случаев (например, поздняя диагностика гипотиреоза, тяжелые сопутствующие заболевания с сердечно-сосудистой и дыхательной недостаточностью) целесообразнее начать с дозы 10-15 мкг/сут, повышая ее каждые 3-5 дней до полной терапевтической
Контроль терапии осуществляют по уровню ТТГ (ниже 5 мМ е/л) и тироксина (130-150 нМоль/л, не допуская гипертиреоза) О хорошей компенсации гипотиреоза свидетельствует физологическая динамика роста, нормализация «костного возраста», активное поведение ребенка. Лечение при ГТ, за исключением транзиторных форм, проводят пожизненно. В начале лечения дополнительно назначают курсы витаминов А и В12. Нейротрофические препараты (пирацетам, церебролизин, липоцеребрин, энцефабол, пантогам) по назначению невропатолога проводят курсами в течение первых 2 лет лечения, затем по показаниям. Обязательны массаж, гимнастика, занятия с логопедом
Прогноз в отношении нейропсихического развития при ВГ зависит от множества факторов. Исследователи во всех странах сходятся во мнении, что определяющую роль для благоприятного прогноза интеллектуального развития ребенка с ВГ, безусловно, играют сроки начала заместительной терапии левотироксином натрия, хотя ряд авторов указывает, что даже при раннем начале лечения у небольшой части детей те или иные нарушения интеллекта все-таки сохраняются. Крайне важным фактором является адекватность лечения на первом году жизни. Таким образом, за некоторым исключением, все дети с ВГ при раннем и адекватном лечении имеют возможность достичь оптимального интеллектуального развития




Профилактика
1.Взятие на учет и наблюдение за беременными женщинами с неблагополучным анамнезом в отношении заболеваний щитовидной железы или проживающих в эндемичных по зобу территориях.
2.Своевременное выявление детей группы риска по развитию гипотиреоза (дети, рожденные от матерей с патологией щитовидной железы, из эндемичных по зобу регионов страны, подвергшиеся воздействию ионизирующего излучения, с гипоплазией щитовидной железы, страдающие вегетососудистой дистонией).
3. Проведение заместительной терапии при гипотиреозе на протяжении всей жизни.
106. Диффузный токсический зоб или Болезнь Грейвса – системное аутоиммунное заболевание, развивающееся вследствие выработки антител к рецептору тиреотропного гормона (АТ-рТТГ), клинически проявляется поражением щитовидной железы с развитием синдрома тиреотоксикоза в сочетании с экстратиреоидной патологией (офтальмопатия, поражение сердечно-сосудистой системы, сопровождающееся тахикардией и др.)
Этиология ДТЗ — заболевание генетически обусловленное, наследуется сцепленно с системой HLA. Тиреоидстимулирующие антитела конкурируют с ТТГ за рецепторы на тиреоцитах, вызывая гиперплазию щитовидной железы и повышение продукции тиреоидных гормонов. Наряду с тиреоидстимулирующими антителами у больных находят антитела к другим тиреоидным антигенам: тиреоглобулину, компонентам коллоида, микросомальной фракции, нуклеарному компоненту. Провоцирующими факторами у детей считают инфекции, чаще носоглотки (хронический тонзиллит), стресс, возможно, химические, токсические вещества.
Патогенез ДТЗ обусловлен аутоиммунным воспалением щитовидной железы с гиперплазией, гипертрофией и лимфоидной инфильтрацией ее, сопровождающимся избыточной продукцией тиреоидных гормонов. В результате повышается нервно-мышечная возбудимость, теплопродукция, увеличивается скорость утилизации глюкозы, потребление кислорода тканями, глюконеогенез, липолиз. Усиление анаболических процессов сочетается с усиленным катаболическим эффектом, вследствие чего развиваются дистрофические изменения в миокарде, печени, мышечная слабость, относительная надпочечниковая недостаточность.
Клиника определяется синдромом тиреотоксикоза, для которого характерны: похудение (часто на фоне повышенного аппетита), потливость, тахикардия и ощущение сердцебиения, внутреннее беспокойство, нервозность, дрожание рук, иногда и всего тела, общая и мышечная слабость, быстрая утомляемость. Классическая триада: зоб, тахикардия и экзофтальм, описанная еще К. Базедовым, встречается примерно у 50% пациентов. В ряде случаев при БГ на первое место могут выходить проявления эндокринной офтальмопатии (выраженный экзофтальм, иногда имеющий несимметричный характер, диплопия при взгляде в одну из сторон или вверх, слезотечение, ощущение «песка в глазах», отечность век). Наличие у пациента выраженной офтальмопатии позволяет практически безошибочно установить этиологический диагноз, поскольку среди заболеваний, протекающих с тиреотоксикозом, эндокринная офтальмопатия сочетается только с БГ.
Диагноз
1.повышение уровней тиреоидных гормонов в крови (Т3 — более 3 нмоль/л, Т4 — более 200 нмоль/л, причем иногда повышен только Т3) при снижении ТТГ обычно ниже 0,1 мкЕД/мл.
2. повышенные уровни тиреоглобулина и тиреоидстимулирующих иммуноглобулинов в крови, коррелирующие со степенью тяжести заболевания
3. УЗИ - диффузное увеличение ЩЖ, характерное для большинства аутоиммунных заболеваниий щитовидной железы; снижение эхогенности щитовидной железы; усиление кровотока в щитовидной железе.
Дифференциальная диагностика
Дифференциальный диагноз Дифференциальный диагноз ДТЗ проводят с другими, сравнительно редко встречающимися в детском возрасте заболеваниями, сопровождающимися гипертиреозом: токсической аденомой щитовидной железы (болезнь Пламмера), гиперфункционирующим раком щитовидной железы или опухолями другой локализации, продуцирующими тиреоидные гормоны, ТТГ-продуцирующей аденомой гипофиза. У детей, получающих тиреоидине препараты, возможен иатрогенный гипертиреоз. У новорожденных от матерей с ДТЗ или аутоиммунным тиреоидитом бывает транзиторный тиреотоксикоз и классический ДТЗ, что определяют по длительности течения болезни.
Иногда сопровождаются гипертиреозом острый и подострый тиреоидиты, имеющие типичную клинику с быстрым, в течение нескольких часов, увеличением щитовидной железы, болезненностью, иногда гипертермией, лабораторными признаками воспаления.
У детей с эутиреоидной гиперплазией щитовидной железы или аутоиммунным тиреоидитом в сочетании с вегетососудистой дистонией имеются некоторые симптомы, общие с тиреотоксикозом, однако при вегетососудистой дистонии тахикардия проходит во время сна и в покое, эмоциональное возбуждение имеет непостоянный характер, и обычно повышено как систолическое, так и диастолическое АД.
Особенно труден дифференциальный диагноз ДТЗ и аутоиммунного тиреоидита (АИТ) в гипертиреоидной фазе. В отличие от ДТЗ, при АИТ щитовидная железа неравномерной плотности, нередко бугристая, с множественными узлами. Гипертиреоз имеет более легкое течение, хорошо поддается консервативному лечению, может пройти спонтанно. Гипертиреоидную стадию АИТ обычно диагностируют в тех случаях, когда гипертиреоз развился у больного, наблюдавшегося более одного года с эутиреоидной или гипотиреоидной стадией АИТ
Лечение: Существует три основных метода лечения
1.Медикаментозная терапия антитиреоидными средствами (метимазол, карбимазол, пропилтиоурацил) начинают с терапии антитиреидными средствами. У детей препаратом выбора служит – тиамазол (торговые названия препарата: мерказолил, метизол, тирозол, метотирин, тапазол, тикапзол, тимидазол). Целью лечения является достижение ремиссии заболевания, стойкая нормализация уровней тиреидных гормонов. Терапия тиамазолом должна продолжаться от 1 до 2 лет, иногда и более
Дети до 1 года - 1,25 мг/сутки; от 1 до 5 лет – 2,5-5,0 мг/сутки; от 5 до 10 лет – 5-10 мг/сутки; от 10 до 18 лет – 10-20 мг/сутки.
2.Хирургическое лечение (тиреоидэктомия) должна быть выбрана в случаях, когда: медикаментозная терапия не привела к стойкой ремиссии; имеются серьезные побочные эффекты при применении медикаментозных средств; необходимо радикальное лечение заболевания; ребенок слишком мал для терапии 131I;
3.Терапия радиоактивным йодом ( 131I) при правильном применении является эффективным средством лечения детей с БГ. Терапии 131I необходимо избегать у очень маленьких детей (младше 5 лет). Терапия 131I вполне допустима у пациентов в возрасте от 5 до 10 лет, если расчетная назначенная активность 131I составляет менее 10 мКи. Возможны ситуации, позволяющие применять 131I терапию и у очень маленьких детей, например развитие серьезных побочных реакций на антитиреоидные средства, недостаток хирургического опыта, или если пациент не является подходящим кандидатом для операции по каким-либо другим причинам
Диспансерное наблюдеие:
-Легкие формы лечат амбулаторно, прочие- в стационаре.
-наблюдение эндокринолога при амбулаторном лечении 2 р/м, 1 р мес после выписки, 1 раз в квартал после утранения тиреотоксикоза
-проводят термометрию, подсчет пульса, АД, размер шеи,анализ крови, Т4,Т3, ТТГ,холестерин и гликемия
-снятие с учета возможно через 3 года после лечения или 2 после операции
Профилактика: предупреждение инфекций,стрессов, избыточной инсоляции, любых втдов излучсений.
Течение: Показателем тяжести гипертиреоза принято считать степень нарушения сердечно-сосудистой системы: стойкая тахикардия в пределах 20% от средних возрастных показателей числа сердечных сокращений характеризует легкую (I) степень тиреотоксикоза, 20-50% — среднетяжелую (II), свыше 50% — тяжелую (III).
Прогноз При правильно проводимой консервативной терапии у 70-80% больных удается достигнуть ремиссии в течение 25 лет . Послеоперационный или возникший после лучевой терапии гипотиреоз требует пожизненной заместительной терапии.
107.Тиреотоксический криз: – тяжелое осложнение тиреотоксикоза, являющееся следствием избыточного выделения тиреоидных гормонов, а симптомы тиреотоксикоза возрастают до степени, угрожающей жизни. ТК может оказаться не только следствием нелеченного тиреотоксикоза, но и стать проявлением неверной тактики его лечения
Этиология:оперативное вмешательство на щитовидной железе; использование радиоактивного йода при тяжелых проявлениях тиреотоксикоза; стрессовые ситуации и физические перегрузки; беременность и роды; интеркуррентные заболевания
Патогенез развития ТК включает:
п овышение
количества свободных фракций тиреоидных
гормонов нарушение связывания
тиреоидных гормонов повышение
чувствительности организма к катехоламинам
развитие острой надпочечниковой
недостаточности.
овышение
количества свободных фракций тиреоидных
гормонов нарушение связывания
тиреоидных гормонов повышение
чувствительности организма к катехоламинам
развитие острой надпочечниковой
недостаточности.
Диагностика. ТК может быть установлен только при подтвержденном диагнозе тиреотоксикоза любой этиологии. Подтверждение тиреотоксикоза включает супрессированный уровень ТТГ и повышение уровней свободных фракций Т4 и Т3.
Для оценки выраженности тяжелых проявлений тиреотоксикоза и ТК используется шкала Burch-Wartofsky
С учетом выраженности проявлений тиреотоксикоза по шкале – BurchWartofsky состояние пациента оценено в 40 баллов - «высокий риск развития ТК». Менее 25 баллов – ТК маловероятен; 25-44 балла – высокий риск развития ТК; более 45 баллов –ТК

Лечение тиреотоксикоза с высоким риском развития ТК или развернутой формы ТК должно быть многокомпонентным и включающим терапию, направленную на:
- контроль повышения активности адренергической системы (блокаторы β-адренорецепторов);
- снижение синтеза тиреоидных гормонов (тионамиды);
-снижение реализации действия тиреоидных гормонов (раствор неорганического йода);
- блокаду периферической конверсии T4 в T3 (йодсодержащие рентгеноконтрастные агенты, ГКС, пропранолол, пропилурацил (незарегистрирован в РБ);
- уменьшение энтерогепатического рециркулирования тиреоидныхгормонов (секвестранты желчных кислот )
- симптоматическую терапию (охлаждение с помощью ацетаминофенаи охлаждающих покрывал, регидратацию, ИВЛ, седативные агенты и др.);
- борьбу с нарушениями сердечного ритма и надпочечниковой недостаточностью;
- противосудорожную терапию.
- Для предотвращения сосудистого коллапса и шока пациентам с ТК проводится возмещение потерянной жидкости (вследствие гипертермии, потоотделения, рвоты, диареи) в количестве, как правило, 3–5 л в сутки (изотонический солевой раствор).
Прогноз и профилактика При правильном лечении острое состояние удается устранить в течение 3-5 дней. В стабильном периоде требуется постоянный контроль уровня гормонов. Ключевым моментом в профилактике тиреотоксического криза является правильная подготовка пациентов к процедуре хирургического вмешательства и к лечению радиоактивным йодом: при помощи медикаментозной терапии должно быть достигнуто относительно стабильное состояние эутиреоза – нормальной концентрации гормонов щитовидной железы в крови. Помимо этого всем пациентам необходим регулярный контроль врача, внеплановые консультации и обследования при развитии инфекционного заболевания, после травм, перед операциями
108. Эндемический (йоддефицитный) зоб— заболевание, встречающееся в местности с низким содержанием йода в воде, почве, растениях, продуктах и характеризующееся увеличением щитовидной железы. По данным ВОЗ, в мире насчитывают более 200 млн больных. Заболевание обычно распространено во многих регионах, удаленных от моря (Альпы, Алтай, Гималаи, Кавказ,. Кордильеры, Карпаты, Тянь-Шань, Центральная Африка, Южная Америка, Восточная Европа). В России эндемическими по зобу районами являются центральные области, Забайкалье, Урал, Сибирь, Дальний Восток. Местность считают эндемичной, если частота зоба по данным УЗИ превышает 5%, а медианная концентрация йода в моче у детей препубертатного возраста (6-12 лет) составляет менее 100 мкг/л.
Этиология
1.Основной причиной эндемического зоба считают недостаточность йода во внешней среде. Помимо йодной недостаточности в развитии эндемического зоба имеют значение поступление в организм зобогенных веществ (тиоцинаты, тиооксизолидоны), йода в недоступной для всасывания форме, пониженное содержание кобальта, меди, цинка, молибдена и других микроэлементов, заболевания желудочно-кишечного тракта, печени, почек, гельминтозы или другие состояния с нарушением всасывания или усиленной экскрецией йода.
2. Вторая группа причин эндемического зоба связана с высокой распространенностью наследственных дефектов захвата йода, «утечкой» йода, нарушением интратиреоидного обмена йода. Так же как при простом зобе, эти нарушения приводят к компенсаторной гиперплазии щитовидной железы, усиливающейся при недостаточности экзогенного йодида.
Патогенез При умеренной нехватке йода потребности организма могут быть удовлетворены при ускорении синтеза тиреоидных гормонов при компенсаторной гиперплазии и гипертрофии щитовидной железы (паренхиматозный зоб), что характерно для детей и подростков. Кроме того, включается целый ряд механизмов адаптации: повышение тиреоидного клиренса неорганического йода, снижение синтеза тиреоглобулина, увеличение синтеза Т3 при снижении Т4, повышение образования в тканях реверсивного Т3 из Т4. С возрастом при длительно сохраняющейся недостаточности йода формируется коллоидный или коллоидно-узловой зоб с высокой вероятностью развития гипотиреоза. Эти изменения наступают тем раньше, чем выше степень йодной недостаточности, и в местности с тяжелой зобной эндемией значительно повышена частота врожденного зоба с гипотиреозом (эндемический кретинизм).
Клиника: При эндемическом зобе, происходит гиперплазия – увеличение щитовидной железы. Это объясняется проявлением реакции организма на пониженную концентрацию йода, а так же вызванный дефицит тиреоидных гормонов. Зачастую совместно с зобом развивается заболевание – гипотиреоз. Организм, наращивая массу щитовидки, пытается компенсировать недостаточное содержание тиреоидных гормонов, благодаря чему возникают следующие симптомы зоба: Головные боли; Дискомфорт в районе сердца; Малая выносливость физических нагрузок; Общая слабость.
Такие симптомы могут возникать, даже на стадии развития заболевания, когда размеры щитовидной железы в норме, а уровень тиреоидных гормонов практически не изменён.
В дальнейшем по мере роста железы, возникают новые симптомы этого заболевания: Приступы удушья; Сухой кашель; Трудности при глотании и дыхании; Ощущения удушья в районе шеи.
Симптомы эндемического зоба вследствие протекания более тяжёлой стадии заболевания, проявляются патологией сердца. Этой патологии характерно гиперфункция и выраженное расширение правого желудочка и предсердия. Среди осложнений эндемического зоба, которые могут возникнуть стоит выделить следующие:Злокачественное перерождение;Кровоизлияние щитовидной железы;Подострый и острый тиреоидиты.
У детей симптомы данного заболевания имеют наиболее интенсивную выраженность. Заболевание чаще всего осложняется в детском возрасте развитием эндемического кретинизма: расстройством ЦНС, задержкой физического и интеллектуального развития.
Диагноз анализ клинических и анамнестических (проживание в эндемической по зобу местности) данных. Для определения объема и структуры щитовидной железы проводят ультрасонографию, а с целью уточнения функции — исследование тиреоидных гормонов и ТТГ в крови. Типичным является умеренное снижение уровня Т4 при повышении Т3 в крови. Содержание ТТГ в крови может быть нормальным или повышенным.
Л ечение:
Эндемический зоб Калия йодид
(йодамарин)200мкг в день 6 мес
ечение:
Эндемический зоб Калия йодид
(йодамарин)200мкг в день 6 мес
Нормализация размеров отсутствие динамики
Йодамарин 50-100 мкг L-тироксин до нормализации
Диф.Диагноз

Профилактика:
Массовая – йодированная соль
Групповая- в группах риска (допубертат 50мкг, пубертат 100мкг, беременные 250мкг)
Индивидуальая решается на приеме у врача
Пораженность населения эндемическим зобом в Башкортостане составляет 30%, что расценивается как тяжелая степень эндемии. В целях профилактики необходимо употребление продукции, обогащенной йодом. Согласно рекомендациям ВОЗ в пищевые продукты добавляется от 10 до 30% суточной нормы йода, в частности йодированная соль содержит йод в количестве 40±15 мкг/г.
Источником йода для человека являются: йодированная соль*, йодированная вода, хлеб, кондитерские изделия, молоко и молочнокислые продукты; богатые йодом продукты моря: морская капуста, рыба, гребешки, крабы и т.п.; специальные продукты для беременных и кормящих женщин: молочные напитки, каши, обогащенные йодом; а так же адаптированные молочные смеси для детей.
109. Аутоиммунный тиреодит (хронический лимфоцитарный тиреоидит, тиреоидит Хашимото) как и диффузный токсический зоб, относят к типичным органоспецифическим аутоиммунным заболеваниям. Он сопровождается лимфоидной инфильтрацией щитовидной железы, наличием специфических антител к нормальным антигенам щитовидной железы и нарушением ее функции. Частота АИТ среди школьников достигает 1%, девочки болеют в 4-7 раз чаще мальчиков.
Этиология 1) факторы внешней среды, 2) факторы внутренней среды Предрасположенность к АИТ имеют лица с носительством антигенов гистосовместимости НLА, 3) другие заболевания, при которых может также развиваться аутоиммунный тиреоидит.
Патогенез. Заболевание обусловлено частичным дефектом иммунологического контроля – дефицитом Тлимфоцитов – супрессоров, в связи с чем происходит выживание запрещенного (форбидного) клона Тлимфоцитов. Взаимодействие форбидного клона Тлимфоцитов с антигенами запускают иммунный процесс по типу гиперчувствительности замедленного типа, выделяются медиаторы воспаления – лимфокины, фактор некроза опухолей и другие цитотоксические вещества.
Т-лимфоциты–хелперы воздействуют на В-лимфоциты, которые превращаются в плазматические клетки и образуют антитела к тиреоглобулину и микросомальной фракции (ТПО). Антитела на поверхности клеток фолликулярного эпителия, объединяясь с Т-лимфоцитами-киллерами, оказывают цитотоксическое действие на гормонпродуцирующие клетки щитовидной железы, вызывают их деструкцию, приводя к снижению функции щитовидной железы, снижают секрецию Т3, Т4 и повышают ТТГ, что приводит к увеличению щитовидной железы – зобу (гипертрофическая форма АИТ)
Атрофическую форму АИТ связывают с блокирующими антителами к рецептору ТТГ. Для гипертрофической формы АИТ характерна диффузная или очаговая инфильтрация лимфоцитами и плазматическими клетками с образованием лимфоидных фолликулов и фиброз различной степени. При атрофической форме АИТ выражен фиброз, атрофия эпителиальных клеток и лимфоидная инфильтрация.
Клиника АИТ может начинаться в любом возрасте, однако наиболее вероятно в 3-6 лет, но диагностируют чаще у подростков или взрослых, в том числе в послеродовом периоде (послеродовый тиреоидит). Начальная стадия характеризуется постепенным диффузным увеличением щитовидной железы, причем у большинства детей не сопровождается болезненностью или другими жалобами. В момент обращения к врачу щитовидная железа обычно диффузно увеличена, имеет неоднородную плотную консистенцию, неровную, бугристую или крупнозернистую поверхность. У некоторых больных имеются симптомы сдавления полых органов шеи, дискомфорт при глотании, периодически — легкая болезненность при пальпации с иррадиацией в ухо и боковые отделы шеи. Клинически чаще можно определить эутиреоидное состояние, иногда симптомы гипотиреоза или гипертиреоза. Атрофические формы АИТ диагностируют при развитии клинической картины гипотиреоза
Диагностика основана на анализе клинических и анамнестических данных.
Ультрасонография щитовидной железы: диффузное ее увеличение, негомогенность стромы, наличие множественных мелких гипоэхогенных участков, расположенных обычно неравномерно в перешейке и обеих долях
В крови значительно повышено содержание антитиреоидных антител, причем их титры обычно нарастают в динамике. Повышен уровень иммуноглобулинов М и G, стойкий лимфоцитоз. Исследование уровней тиреоидных гормонов и ТТГ в крови не требуется для диагноза, но обязательно для назначения лечения. Иногда бывает субклинический гипотиреоз (повышение ТТГ при нормальных уровнях Т3 и Т4) или явный первичный гипотиреоз (снижение Т3 и Т4 при повышенных уровнях ТТГ в крови); иногда выявляют гипертиреоз
В сомнительных случаях, а при наличии узлов обязательно, проводят пункционную биопсию щитовидной железы под контролем ультрасонографии с последующим гистологическим исследованием биоптата
Дифференциальный диагноз проводят с простым зобом, эндемическим зобом, при наличии гипертиреоза — диффузным токсическим зобом.
Дифференциальный диагноз с подострым вирусным тиреоидитом основан на характерной клинической картине: начало заболевания через 7-14 дней после перенесенной вирусной инфекции, быстрое, в течение нескольких часов увеличение щитовидной железы, сопровождающееся гипертермией, болезненностью, иногда гиперемией кожи над щитовидной железой. Характерно значительное повышение СОЭ (50-60 мм/час) при нормальном содержании лейкоцитов в крови. Антитиреоидные антитела появляются на 2-й неделе заболевания, и их титры снижаются по мере выздоровления в течение 6-18 мес.
Острые бактериальные, грибковые, специфические (туберкулезный, бруцеллезный, сифилитический и др.) тиреоидиты дифференцируют по типичной клинике, данным клинического анализа крови, пункционной биопсии, микроскопии и посевам материала биопсии.
Лечение. Показания к лечени: дети имеющие явное снижение тиреоидной функции(норм ттг, повыш т4) –гипотериоз, дети с субклиническим гипотериозом(норм т4 и повыш ттг), дети с увелиц ЩЖ
Левотироксин – критерием адекватности считать достижение норм показателя ттг и стойкое его сохранение.
Диспансеризация:

Прогноз аутоиммунного тиреоидита у детей благоприятный. Если вовремя начать лечение, то удастся полностью вылечить заболевание и не допустить в дальнейшем рецидивов. Подострая форма заболевания проходит часто самостоятельно, не провоцирует тяжелых последствий для здоровья и не влияет на дальнейшую работу железы.
110.Низкорослость – это отставание длины и тела от средних показателей для этго возраста и пола более, чем на 2 стандартных отклонения.
Классификация причин низкорослости
1. Первичные нарушения роста:
- Скелетные дисплазии.
- Хромосомные нарушения, сочетающиеся с низкорослостью.
- Внутриутробная задержка роста.
2. Вторичные нарушения роста:
-Хронические заболевания специфических систем
-Эндокринные заболевания:
-Голодание.
-Ятрогенные нарушения.
-Психосоциальный нанизм.
3. Конституциональная задержка роста и пубертата.
4. Семейная низкорослость.
5. Дефицит гормона роста.
-Врожденные формы
-Приобретенные формы
Первичные нарушения роста
Скелетные дисплазии представлены гетерогенной группой заболеваний, которые имеют наследственную предрасположенность и характеризуются выраженными дефектами развития хрящевой и/или костной ткани с формированием диспропорциональной низкорослости и изменением размера и/или формы различных частей скелета. Частота встречаемости скелетных дисплазий 30–45 случаев на 100 000 новорожденных. Известно более 100 форм заболевания. Для многих форм костных дисплазий генных дефектов, основными критериями при постановке диагноза остаются клинические и рентгенологические признаки заболевания. В зависимости от характера нарушений пропорции тела ребенка выделяют скелетные дисплазии с укорочением конечностей и с укорочением туловища.
Ахондроплазия. Тип наследования аутосомно-доминантный с полной пенетрантностью. Типичные проявления заболевания позволяют установить диагноз при рождении ребенка. Характерными признаками являются диспропорциональное телосложение с укорочением проксимальных отделов конечностей, относительно длинное туловище, короткие пальцы рук, кисть в форме «трезубца», поясничный лордоз. Определяется повышенное разгибание большинства суставов, особенно коленных; выражена варусная деформация голеней, мегалоцефалия, курносость. До 2 лет жизни больного задержка роста может не наблюдаться, позднее отмечается выраженная низкорослость. Часто наблюдаемое отставание в моторном развитии ребенка обусловлено мышечной гипотонией. Интеллект сохранен. Средний конечный рост мужчин составляет 131 см, женщин — 124 см.
Хромосомные нарушения Синдром Дауна В основе заболевания лежит наличие дополнительной 21-й хромосомы или части ее длинного плеча, включающей фрагмент q22.1. Частота встречаемости 1 случай на 600 живых новорожденных. При рождении масса тела больного на 500 г меньше и рост на 2–3 см ниже средне популяционных норм. Низкорослость проявляется постнатально, скорость роста резко замедляется после 6–9 месяцев жизни, в 3 года рост ребенка ниже 3-й перцентили для данного пола. Наблюдается отставание костного возраста, задержка и низкий пубертатный скачок роста. Типичными признаками заболевания являются задержка умственного развития, мышечная гипотония, плоское лицо, монголоидный разрез глаз, зубные аномалии, короткие конечности, брахимезофалангия. Рост мужчин — 135–170 см, женщин — 127–158 см. У части детей выявляется соматотропная недостаточность, гипотиреоз.
Внутриутробная задержка . рост и масса ребенка при рождении отстают от нормальных показателей для соответствующего гестационного возраста более чем на 2 стандартных отклонения. ВЗР может быть составляющей одного из наследственных синдромов или быть самостоятельным заболеванием. Причины ВЗР: Многоплодная беременность. Нарушение питания матери (недоедание). Конституция матери.. Экстрагенитальные заболевания матери (артериальная гипертензия, сахарный диабет). Вредные факторы воздействия (курение, алкоголизм, интоксикация). Плацентарные пороки. Генетические пороки плода
Вторичные нарушения роста
Эндокринные заболевания Гипотиреоз Замедление скорости роста характерный признак снижения функции щитовидной железы. Тиреоидные гормоны оказывают непосредственное влияние на гипертрофическую зону эпифизарной ростовой пластинки и активно участвуют в регуляции секреции ГР. Врожденный гипотиреоз как причина низкорослости у детей, благодаря проводимому скринингу и своевременному адекватному лечению, встречается редко. Основной причиной задержки роста в детском возрасте может являться длительно не диагностированный приобретенный гипотиреоз (например, при аутоиммунном тироидите).
Голодание Неадекватное возрастным потребностям поступление в организм ребенка белков, микроэлементов (цинк, железо) и витаминов, недостаточная калорийность пищи являются частыми причинами задержки роста. При низкорослости вследствие белкового/калорийного голодания отмечается повышение базального и/или стимулированного уровней ГР. Синтез ИФР-1 и ИФРСБ-3 снижен. Эти изменения ростовых параметров являются проявлением адаптационного перераспределения белковых резервов, направленных на поддержание существования организма.
Конституциональная задержка роста и пубертата характерно отсроченное пубертатное ускорение роста, обусловленное более поздним началом полового развития. Так мальчики достигают 2-й стадии по Таннеру в возрасте 13,8 лет, девочки — в 13,3 года. Сроки начала пубертата и сопутствующее ему ускорение роста зависят от степени отставания костного возраста. Как правило, в семейном анамнезе имеется указание на задержку полового развития у отца или родственников по отцовской линии.
Дефицит гормона роста
Врожденный (например наследственный гипопитуитаризм см. след вопрос)
Приобретенный деф гормона ростаОпухоли гипоталамо-гипофизарной системы Клиника приобретенной недостаточности ГР аналогична таковой у детей с врожденными формами заболевания, однако для нее характерны некоторые особенности. Отсутствуют признаки, обусловленные перинатальным дефицитом ГР: недоразвитие костей лицевого скелета, акромикрия, микрогенитализм. Нет гипогликемических реакций. Характерно острое начало заболевания, 24 т. е. резкой задержки роста, при отсутствии каких-либо прогрессирующих отклонений роста от перцентильной кривой с рождения. Как правило, рост ребенка длительное время соответствует генетическому ростовому коридору и только на фоне манифестации основного заболевания происходит его замедление
111. Гипопитуитаризм (гипофизарный нанизм, гипоталамический нанизм) — заболевание, обусловленное недостаточностью соматотропного гормона (СТГ), проявляющееся у детей значительной задержкой роста. Абсолютная или частичная недостаточность СТГ может развиваться не только вследствие патологии гипофиза, но и при нарушении гипоталамической регуляции его секреции, дефектах биосинтеза СТГ, снижении чувствительности периферических тканей к гормону. У части больных обнаруживают изолированный (абсолютный или частичный) дефицит СТГ, но в большинстве случаев имеется недостаточность и других гормонов гипофиза в разных сочетаниях, что приводит к формированию множества клинических вариантов заболевания с различной комбинацией эндокринных и обменных нарушений.
Этиология Наиболее частой причиной гипопитуитаризма считают деструктивные изменения в гипоталамусе или гипофизе, возникающие в результате травмы, в том числе родовой, кровоизлияния, гипоксии
Классификация по прич
I. Врожденный гипопитуитаризм:
1. Наследственный:
- Изолированный дефицит СТГ:
-Множественный дефицит гормонов аденогипофиза:
2. Идиопатический дефицит СТГ'Р.
3. Дефекты развития гипоталамо'гипофизарной системы:Патология срединной трубки, Дисгенез гипофиза
II. Приобретенный : Опухоли гипоталамуса и гипофиза, Опухоли других отделов мозга, Травмы, Инфекции, Супраселлярные арахноидальные кисты, гидроцефалия ,Сосудистая патология, Облучение головы и шеи, Токсические последствия химиотерапии, Инфильтративные болезни
Патогенез Недостаточность СТГ приводит к снижению синтеза в печени, почках и других органах соматомедина С или инсулиноподобного фактора роста I (ИРФ I), который непосредственно стимулирует синтез белка и деление хрящевых клеток, рост соединительной ткани, связок, суставов, кожи, недифференцированных клеток крови, периферических нервов и др. В результате резко задерживается рост скелета, мышц, внутренних органов, снижается их функция. Снижается утилизация глюкозы, тормозится липолиз, глюконеогенез. Снижение секреции гонадотропинов, ТТГ, АКТГ или соответствующих рилизинг-гормонов приводит к снижению функции щитовидной железы, надпочечников, гонад.
Клиника. Врожденный гипопитуитаризм в раннем возрасте не специфична. Родители, как правило, среднего роста. Рост ребенка при рождении обычно средний, иногда умеренно задержан. У мальчиков могут быть уменьшены размеры полового члена, а у девочек — гипоплазия малых половых губ. В периоде новорожденности характерны апноэ, судороги, гипотония, гипогликемии, пролонгированная желтуха.
Скорость роста у детей при выраженной недостаточности СТГ обычно ниже 3 центилей и не превышает 2-3 см в год.
Существенное отставание длины тела и скорости роста диагностируют обычно через 2-3 года от начала заболевания, то есть при врожденном гипопитуитаризме задержка роста становится очевидной через несколько лет после рождения, хотя снижение скорости роста выявляют раньше. Типичным является отставание «костного возраста» от паспортного и соответствие «возрасту по длине тела». При абсолютном дефиците СТГ ребенок имеет типичный внешний вид: голова круглая, лицо короткое и широкое . Задержано закрытие родничков черепа иногда до 2-3 лет. Лобная кость выступает вперед, корень носа седловидной формы, западает, нос маленький, резко выражены носогубные складки. Нижняя челюсть и подбородок недоразвиты, задерживается прорезывание зубов. Шея короткая, голос высокий, маленькие кисти и стопы. Телосложение инфантильное, кожа суховатая, дряблая, с желтоватым оттенком. Подкожный жировой слой часто развит избыточно, особенно у детей старшего возраста, распределен по женскому типу. Мышцы развиты слабо.Больные имеют нормальный интеллект, но обладают замкнутым, стеснительным или агрессивным характером.
Приобретенные формы гипофункции гипофиза развиваются в любом возрасте, чаще в периоде новорожденности, и тогда ведущим симптомом может быть неонатальная гипогликемия. Длина тела ребенка при рождении средняя и низкорослость выявляют через 1 -2 года после начала заболевания. Сохраняются инфантильные пропорции тела, возникает астения, снижаются прибавки массы тела, повышается чувствительность к холоду. Половое созревание не наступает, а если уже началось — регрессирует. Иногда появляются симптомы несахарного диабета. Если причина заболевания — опухоль, нарастают неврологические симптомы: головная боль,рвота, нарушение зрения, сна. Примерно у половины детей задержка роста предшествует появлению неврологических расстройств.
Диагноз гипопитуитаризма основан на анализе анамнестических и клинических данных. Задержка роста более 3 центиля со значительным, пропорциональным степени низкорослости, отставанием «костного возраста», особенно в сочетании с аномалиями развития средней части лица с большой вероятностью указывают на гипопитуитаризм.
1.Определение СТГ в крови натощак, как и в любой случайной пробе крови, диагностического значения не имеет. В соответствии с современными требованиями, диагноз гипопитуитаризма должен быть подтвержден при помощи минимум двух медикаментозных тестов — с инсулином, клонидином, дофамином, аргинином
В оценке результатов медикаментозных стимуляционных тестов принято придерживаться следующих нормативов: соматотропная недостаточность считается доказанной, если ни в одной из проб крови в ходе двух стимуляционных тестов уровень СТГ не превышает 7,0 нг/мл. Показатели от 7 до 10 нг/мл принято считать сомнительными или свидетельствующими о частичной недостаточности СТГ и требующими перепроверки, а уровень СТГ в любой пробе крови 10 нг/мл и выше исключает соматотропную недостаточность.
2.Исследование базальных уровней И РФ -I и ИРФСБ-3 в крови, особенно у детей старше 5 лет, можно использовать в амбулаторных условиях в качестве критерия для отбора детей с подозрением на соматотропную недостаточность для проведения стимуляционных тестов.
3.базальные уровни ТТГ, свободного Т4 (или общих Т3 и Т4), ПРЛ, АКТГ, кортизола, а у детей пубертатного возраста — Л Г, ФСГ и половых гормонов в крови. Следует помнить, что при некомпенсированном гипотиреозе и гипогонадизме (у подростков) возможно выявление сниженных уровней СТГ и в отсутствие гипопитуитаризма, поэтому перед проведением функциональных проб рекомендуют начать заместительную терапию соответствующими гормонами.

Лечение
Заместительную терапию гормоном роста проводят при доказанной недостаточности СТГ с момента подтверждения диагноза в дозе 0,07-0,1 ЕД/кг в сутки (0,5-0,7 МЕ/кг в неделю), вводя препарат ежедневно или 6 раз в неделю подкожно, длительно, до закрытия зон роста или достижения социально приемлемого роста.
При недостаточности ТТГ назначают тиреоидные гормоны в полных заместительных дозах. Наличие надпочечниковой недостаточности вследствие дефицита АКТГ — показание для назначения кортизона в дозе не более 10-15 мг/м2 в сутки. Заместительную терапию половыми гормонами начинают обычно при костном возрасте более 12 лет.
Прогноз у большей части больных при рано начатом (до 5-7 лет) и правильно проводимом лечении вполне благоприятный — дети достигают среднего роста 160-175 см. Однако им целесообразно проводить пожизненную гормональную заместительную терапию, позволяющую компенсировать нарушения обмена и добиться улучшения качества жизни. При начале лечения после 1 0-12 лет, а также отсутствии регулярной заместительной терапии, часто не удается достичь социально приемлемого роста. Прогноз для жизни становится серьезным при наличии интракраниальных опухолей.
112. Гигантизм Гигантизм – патологическая высокорослость, обусловленная чрезмерной выработкой гормона роста (соматотропного гормона) передней долей гипофиза и проявляющаяся уже в детском возрасте. Наблюдается увеличение роста свыше 3 ц , непропорциональность телосложения с преимущественным удлинением конечностей, при этом голова кажется очень маленькой. У больных наблюдается расстройство физического и психического состояния, половой функции. При гигантизме трудоспособность ограничена, высок риск бесплодия. Основным диагностическим критерием гигантизма, помимо яркой клинической картины, является выявление повышения СТГ в крови.
Причины гигантизма
Основополагающей причиной гигантизма является чрезмерная выработка соматотропина, которую в свою очередь могут спровоцировать следующие патологии:
интоксикация (повышенный уровень токсинов в организме);
новообразования аденогипофиза;
черепно-мозговые травмы;
нейроинфекции — инфекционные (бактериальные или вирусные) заболевания центральной нервной системы, такие как энцефалит, менингит, менингоэнцефалит.
Классификация гигантизма
Согласно современной эндокринологической классификации, выделяют следующие виды гигантизма:
истинный гигантизм, характерным признаком которого является пропорциональное увеличение всех частей тела, при этом психическое и функциональное развитие организма остается в норме;
акромегалический гигантизм (присоединяются признаки акромегалии);
спланхомегалия: при данном виде гигантизма происходит увеличение размеров и массы внутренних органов, в некоторых источниках данное заболевание встречается под названием «гигантизм внутренних органов»;
евнухоидный гигантизм, патология главной особенностью, которой является снижение функциональности или полная дисфункция половых желез. У таких пациентов практически отсутствуют вторичные половые признаки, есть непропорционально удлиненные конечности и открытые зоны роста в суставах;
парциальный или частичный гигантизм сопровождается увеличением отдельных частей тела;
половинный гигантизм характеризуется увеличением тела с одной стороны;
церебральный гигантизм: сопряжен с органическим нарушением со стороны головного мозга и влечет за собой расстройства интеллекта.
113. Гипофизарный гигантизм: этиология, патогенез, клиника, диагностика, дифференциальный диагноз различных форм высокорослости (семейная высокорослость, преждевременное половое развитие, синдром Марфана, синдром Клайнфельтера), лечение, диспансерное наблюдение, прогноз.
Акромегалия – нейроэндокринное заболевание, вызванное хронической избыточной секрецией гормона роста (СТГ) у лиц с законченным физиологическим ростом и характеризующееся патологическим диспропорциональным периостальным ростом костей, хрящей, мягких тканей, внутренних органов, а также нарушением функционального состояния сердечно-сосудистой, легочной системы, периферических эндокринных желез, различных видов метаболизма). Гигантизм – нейроэндокринное заболевание, вызванное хронической избыточной секрецией гормона роста, возникающее у детей и подростков с незаконченным физиологическим ростом, характеризующееся 2 пропорциональным ростом костей скелета в длину, приводящее к значительному увеличению роста субъекта или
Гипофизарный гигантизм — заболевание, в основе которого лежит гиперфункция эозинофильных клеток передней доли гипофиза. Макроскопически гипофиз может быть нормальным или увеличенным, микроскопически не обнаруживают никакой аномалии, определяют относительное преобладание или аденому эозинофильных клеток. При развитии опухоли до закрытия эпифизарных зон роста развивается гигантизм, позже — акромегалия. В детском возрасте наблюдается, как правило, гигантизм, однако возможны и гипофизарные гиганты с акромегалоидными признаками. Усиленный рост организма начинается обычно в пубертатном периоде (10 — 14 лет), реже в раннем детском возрасте. Заболевание встречается преимущественно у мужского пола.
Этиология. Основным этиологическим фактором считают эозинофильную аденому или гиперплазию эозинофильных клеток. Инфекционные заболевания, в том числе энцефалит, также могут вызвать эту патологию. Одним из дополнительных факторов в генезе заболевания является свойство костей усиленно реагировать на стимуляцию гормоном роста. Наследственный фактор! Клинические признаки. Принято различать 2 формы заболевания. В одних случаях усиленный рост происходит постоянно, в других — толчками, с наличием периодов относительного покоя. Сначала рост пропорциональный. Затем туловище и конечности удлиняются, кисти и стопы достигают громадных размеров, появляются акромегалоидные признаки (утолщение костей черепа, огрубение черт лица, прогнатизм, увеличение промежутков между зубами), кифоз из-за чрезмерного роста позвонков. Половое развитие вначале бывает нормальное или преждевременное. Отмечаются симптомы гипотиреоза, гипергликемия и гликозурия. По мере роста аденомы и сдавления гипофиза гиперфункция его сменяется гипофункцией. Больные предъявляют жалобы на головную боль и боль в конечностях. Появляется мышечная слабость, снижается функция половых желез, развивается недостаточность функции щитовидной железы, наблюдается сдавление хиазмы зрительных нервов с развитием битемпоральной гемианопсии и слепоты. У детей возможен частичный гигантизм — увеличение какой-либо части тела или органа (отдельных пальцев, кисти, ступни или всей конечности). Частичный гигантизм — чаще врожденная патология. В развитии частичного гигантизма придают значение усиленному росту части тела или органа в связи с ненормальным положением плода в матке или сдавлением органа пуповиной и развитием в нем застойных явлений. Нарушениям в закладке и формировании органов. патологии центральной нервной системы. Мышцы пораженной части тела гипертрофированы, кости увеличены в размерах, выражены явления остеопороза. Диагноз заболевания ставится при наличии гигантского роста (ориентировочно у женщин 190 см, у мужчин — 205 см). О гипофизарном гигантизме свидетельствует пропорциональный рост костей, за исключением непропорционально длинных костей конечностей. Дифференцируют заболевание с арахнодактилией, гипертиреозом, адреногенитальным синдромом, синдромом Олбрайта. Кроме того, о гипофизарном гигантизме свидетельствует гиперфосфатемия и повышение энергетического обмена.
Основные диагностические мероприятия Амбулаторно: - Определение базального уровня СТГ в сыворотке крови 2-3 раза - Определение в крови уровня пролактина. - Определение в крови уровня ИРФ-1 (соматомедина-С) - Компьютерная, либо магнитно-резонансная томография области турецкого седла с контрастированием. - Оценка состояния глазного дна, области перекреста зрительных нервов В стационаре: - ОГТТ с 75 г глюкозы с определением СТГ на 0, 30, 60, 90, 120 мин (проводится только у пациентов, не страдающих сахарным диабетом!). - МРТ или КТ органов грудной клетки и брюшной полости для выявления эктопированной опухоли (при наличии показаний) Физикальное обследование: Основные клинические проявления - Изменение внешности (укрупнение носа, губ, языка, утолщение кожи, увеличение надбровных дуг), увеличение верхней и нижней челюсти, прогнатия, расширение межзубных промежутков – диастема, увеличение конечностей. - Увеличение внутренних органов – спланхномегалия - Себорея, гипергидроз, акне - Признаки объем ного образования хиазмально-селлярной области: головная боль, нарушения полей зрения, парезы черепно-мозговых нервов, гиперпитуитаризм, гиперпролактинемия - Парестезии, артралгии, корешковые и туннельные синдромы - Артериальная гипертензия, кардиомегалия - Нарушение менструального цикла, галакторея, снихение либидо, потенции - Нарушение толерантности к глюкозе, сахарный диабет - Гиперхолестеринемия, гипрертриглицеридемия - Склонность к новообразованиям (полипы ЖКТ, узловой зоб, миома матки) Инструментальные методы исследования: - Наличие аденомы гипофиза при проведении компьютерной, либо магнитно-резонансной томографии с контрастированием. - Наличие эктопированной опухоли, секретирующей СТГ или соматолиберин - Увеличение толщины мягких тканей стопы в области пяточной кости. Норма у мужчин до 21 мм, у женщин до 20 мм. - Изменения на глазном дне и гемианопсия, выявляемые при оценке состояния глазного дна, области перекреста зрительных нервов, периметрии Голдмана. ЛЕЧЕНИЕ При исключении конституционального гигантизма для замедления роста у девушек рекомендуется применять эстрогены. Они ускоряют окостенение эпифизарных хрящей и способны тормозить выделение соматотропного гормона. (эстрадиол-бензоат по 1—6 мг через день внутримышечно или стильбестрол внутрь по 2—10 мг ежедневно). При появлении маточных кровотечений доза эстрогенов удваивается. При подозрении на наличие аденомы гипофиза и признаков прогрессирования заболевания — рентгенотерапия межуточно-гипофизарной областиВ тяжелых случаях акромегалии в результате прогрессирующего роста опухоли, угрозе полной потери зрения — гипофизэктомия.
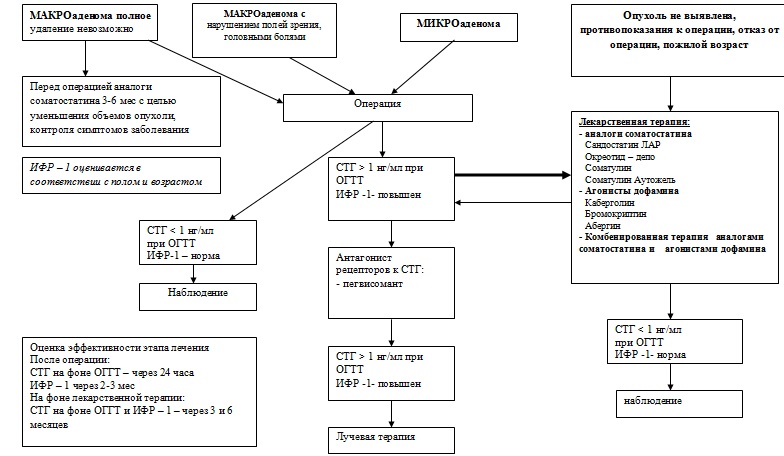
Прогноз. Продолжительность жизни гипофизарных гигантов составляет 30—40 лет.
Дальнейшее ведение: После оперативного лечения и лучевой терапии пациент наблюдается у эндокринолога. Эффективность хирургического вмешательства контролируется определением ИРФ-1(инсулиноподобный ростовой фактор) или проведением ОГТТ с определением СТГ в раннем послеоперационном периоде – на 5-8 сутки и через 3, 6 и 12 месяцев. Один раз в год проводится определение ТТГ, свТ4, кортизола, половых гормонов. Эффективность лучевой терапии с исследованием ИРФ-1 или проведением ОГТТ с определением СТГ оценивается не ранее, чем через 6 месяцев. Эффективность лечения аналогами соматостатина должна контролироваться каждые 3-4 месяца (ИФР-1 или ОГТТ с определением СТГ). У пациентов с медикаментозной ремиссией для определения остаточной функции гипофиза через 1-2 года после начала терапии необходимо прервать лечение на 1 – 2 месяца с контролем ИФР-1. При сохранении нормального уровня ИФР-1 лечение прекращают. Наблюдение нейрохирурга, КТ и МРТ через 6-12 месяцев. Осмотр окулиста 1 раз в год.
Синдром Марфана - дифференцированная форма врожденной соединительнотканной недостаточности, характеризующаяся разнообразными проявлениями скелетной, сердечно-сосудистой и глазной патологии. У больных с синдромом Марфана отмечаются гигантизм, долихостеномелия и арахнодактилия, аневризмы аорты, миопия, эктопия хрусталика, деформация грудины, кифосколиоз, плоскостопие, протрузия вертлужной впадины, эктазия твердой мозговой оболочки. Диагноз синдрома Марфана основан на семейном анамнезе, результатах функционального, офтальмологического, рентгенологического и генетического исследований. Лечение при синдроме Марфана включает консервативную и хирургическую коррекцию сердечно-сосудистых нарушений, поражений скелета и органа зрения. Конституционально-наследственная, или семейная, высокорослость обусловленна генетическими факторами. Она является вариантом нормы и встречается чаще у лиц мужского пола. Семейная высокорослость характеризуется высокими темпами роста, пропорциональным ростом и телосложением, нормальным ростом и массой тела при рождении, костный возраст соответствует хронологическому, умственное развитие в норме. Преждевременное половое развитие – патологическое состояние, характеризующееся появлением признаков полового развития у девочек до 8, у мальчиков – до 9 лет. Синдром Клайнфельтера - хромосомная патология, обусловленная наличием в мужском кариотипе одной или нескольких дополнительных женских половых хромосом. Синдром Клайнфельтера характеризуется первичным гипогонадизмом, маленькими размерами тестикул, бесплодием, гинекомастией, неглубоким снижением интеллекта. Решающая роль в диагностике синдрома Клайнфельтера принадлежит кариотипированию; также проводится анализ фенотипических признаков, определение полового хроматина, экскреции фолликулостимулирующего гормона с мочой, спермограмма и пр. Лечение при синдроме Клайнфельтера включает гормональную терапию, возможно - оперативную коррекцию гинекомастии, однако полное излечение синдрома невозможно.
114. Ожирение: определение, эпидемиология, этиология, патогенез, классификация, клиника, диагностика, дифференциальный диагноз различных форм ожирения, лечение, диспансерное наблюдение, прогноз, профилактика.
Ожирение – избыточные жировые отложения в подкожной клетчатке, органах и тканях. Проявляется увеличением массы тела на 20 и более процентов от средних величин за счет жировой ткани. Этиология. Ожирение относится к многофакторным заболеваниям. Самый частый вид ожирения, связанный с избыточным поступлением калорий в условиях гиподинамии и наследственной предрасположенности – конституционально-экзогенное (простое, идиопатическое) ожирение. Несмотря на то, что конст-экзогенное ожирение является заболеванием с наследственной предрасположенностью, вероятность его развития и степень проявления во многом зависят от образа жизни и характера питания. Избыточные калории, поступившие в организм и не израсходованные им, преобразуются в жир, который накапливается в жировых депо организма (преимущественно в подкожной клетчатке, сальниках, брюшной стенке, внутренних органах и т. д.). Изменения в пищевом поведении происходят в результате нарушения гипоталамо-гипофизарной регуляции, отвечающей за контроль поведенческих реакций. Повышение активности гипоталамо-гипофизарно-адреналовой системы ведет к увеличению продукции АКТГ, скорости секреции кортизола и ускорению его метаболизма. Происходит снижение секреции соматотропного гормона, оказывающего липолитическое действие, развивается гиперинсулинемия, нарушение метаболизма тиреоидных гормонов и чувствительности к ним тканей.
Развитию ожирения способствует ряд факторов:
· малоактивный образ жизни;· генетически обусловленные нарушения ферментативной активности · погрешности в характере и режиме питания · некоторые эндокринные патологии (гипотиреоз, гипогонадизм, инсулинома, болезнь Иценко-Кушинга); · психогенное переедание; · физиологические состояния (лактация, беременность, климакс);· стрессы, недосыпание, прием психотропных и гормональных препаратов (стероидов, инсулина, противозачаточных таблеток) и т. д.
Классификация ожирения По этиологии: простое (конституционально-экзогенное, идиопатическое) — ожирение, связанное с избыточным поступлением калорий в условиях гиподинамии и наследственной предрасположенности; гипоталамическое — ожирение, связанное с наличием и лечением опухолей гипоталамуса и ствола мозга, лучевой терапией опухолей головного мозга и гемобластозов, травмой черепа или инсультом; ожирение при нейроэндокринных заболеваниях (гиперкортицизме, гипотиреозе и др.); ожирение ятрогенное (вызванное длительным приемом глюкокортикоидов, антидепрессантов и других препаратов); моногенное ожирение — вследствие мутаций генов лептина, рецептора лептина, рецепторов меланокортинов 3-го и 4-го типа, проопиомеланокортина, проконвертазы 1-го типа, рецептора нейротрофического фактора — тропомиозинсвязанной киназы B, см. табл.2); синдромальное ожирение (при хромосомных нарушениях, заболеваниях вследствие геномного импринтинга, других генетических синдромах — Прадера— Вилли, хрупкой X-хромосомы, Альстрема, Кохена, Дауна, при псевдогипопаратиреозе и др
классификация степеней ожирения, основанная на определении показателя – индекса массы тела (ИМТ) для лиц от 18 до 65 лет. ИМТ рассчитывается по формуле: вес в кг / рост в метрах в квадрате. По ИМТ выделяют следующие варианты массы тела и риска развития сопутствующих осложнений:· ИМТ от 18,5 до 24,9 (обычный) – соответствует массе тела в норме. ИМТ от 30 и более указывает на наличие ожирения и прямой угрозы здоровью, ожирение делится на 4 степени: при I степени избыточная масса составляет не более 29%, II степень характеризуется превышением массы на 30-40%, III – на 50-99%, при IV(морбидное) степени отмечается увеличение фактической массы тела по сравнению с идеальной в 2 и более раз.
Клиника. Клиническая картина определяется этиопатогенетической формой ожирения. Дебют заболевания чаще всего в возрасте после 5 лет, или в периоде полового созревания. Как правило, ожирение прогрессирует постепенно, на фоне хороших (часто ускоренных) темпов роста. Наличие стрий, фолликулярного кератоза, полифагии, черного акантоза, артериальной гипертензии и др. не всегда коррелирует со степенью ожирения. Характерно наличие избыточной массы тела и ожирения у родственников Гипоталамическое ожирение, развивается после оперативного вмешательства (лучевой терапии). В случае краниофарингиомы для большинства пациентов характерно замедление темпов роста; для глиом симптомы преждевременного полового развития; неврологические жалобы (головные боли, нарушение зрения) зависят от локализации и прогрессии опухоли. При моногенных формах ожирение дебютирует в первые месяцы и годы жизни, для большинства синдромальных форм характерна задержка психомоторного развития.
Диагностика ожирения. Поскольку непосредственно оценить количество жировой ткани в организме сложно, наиболее информативным является определение ИМТ, который рассчитывается как отношение массы тела в килограммах к квадрату роста человека, выраженному в метрах. Доказано, что ИМТ коррелирует с количеством жировой ткани в организме как у взрослых, так и у детей. Критерии избыточной массы тела у детей определяются по данным перцентильных таблиц или стандарных отклонений ИМТ. В них учитывается не только рост, вес, но также пол и возраст ребенка. Это связано с тем, что значение ИМТ у детей меняется с развитием ребенка: от высокого в первый год жизни, сниженного в период раннего детства (2—5 лет) и постепенно увеличивающегося в период полового развития, что в целом отражает динамику жировой ткани. Данные нормативы объединяет общий принцип: перцентили должны быть симметричны относительно медианы (50-й перцентили).
Всем детям с ожирением и избыточной массой тела рекомендуется: 1. проводить измерения роста, SDS роста, веса с расчетом SDS ИМТ, окружности талии, 2. оценивать характер распределения подкожной жировой клетчатки, 3. проводить измерение артериального давления (АД) и оценивать его с учетом пола, возраста и роста, 4. определять наличие и характер стрий, фолликулярного кератоза, , андрогензависимой дермопатии (у девочек – гирсутим, акне, жирная себорея), 5. оценивать стадию полового развития, 6. выявлять специфические фенотипические особенности (характерные для синдромальных форм)
Всем пациентам с ожирением рекомендованы биохимические исследования: липидограмма крови, уровни ферментов печени (АлАТ, АсАТ) в сочетании с ультразвуковым исследованием печени
Дифференциальную диагностику конституционально-экзогенного ожирения следует проводить с генетическими синдромами, ассоциированным с ожирением (синдромальные и моногенные формы), с гипоталамическим ожирением, нейроэндокринным и ятрогенным ожирением. Скрининг на наличие генетических форм ожирения рекомендуется детям с ранним (до 5 лет жизни) ожирением на фоне выраженной гиперфагии, особенно при наличии выраженного ожирения в семейном анамнезе
Дополнительно:
Синдромальные формы ожирения характеризуются ранним дебютом ожирения и его быстрым прогрессированием. Для большинства синдромальных форм характерна задержка нервно-психического развития от умеренной до тяжелой степени выраженности, наличие дисморфических признаков и органоспецифических аномалий развития
Моногенные формы ожирения встречаются крайне редко, отличаются ранним дебютом (с первых месяцев жизни – до 1 года), полифагией. Для большинства пациентов характерно нормальное нервно-психическое развитие (см. табл. 3). Моногенные формы ожирения развиваются из-за мутации одного из генов, кодирующих белки лептинмеланокортиновой системы.
Врожденный дефицит лептина — редкий синдром, характеризующийся морбидным ожирением с первых месяцев жизни на фоне выраженной гиперфагии и низким, неопределяемым уровнем лептина. Для этих пациентов характерен гипогонадотропный гипогонадизм, дефицит гормона роста, гипотиреоз центрального генеза, также дети с дефицитом лептина часто болеют респираторными инфекционными заболеваниями из-за гипофункции Т-клеток
Гипоталамическое ожирение развивается вследствие опухолей гипоталамуса и ствола мозга, лучевой терапии опухолей головного мозга и гемобластозов, перенесенных травм черепа. Отличительной чертой гипоталамического ожирения является выраженная полифагия, развивающаяся непосредственно после проведения лечения и приводящая к стремительному набору веса. Дети и подростки с гипоталамическим ожирением часто предъявляют жалобы на нарушения ритма сна и бодрствования, частые головные боли и другие неврологические и поведенческие нарушения.
Лечение: Изменение образа жизни (диетотерапия-рассказать, что ограничивем, расширение физической активности и коррекция пищевого поведения) у детей и подростков с ожирением или избыточной массой тела, а также членов их семьи составляют основу терапии ожирения и его профилактики. В случае неэффективности модификации образа жизни возможно использование фармакологических средств, список которых у детей и подростков ограничен орлистатом. Бариатрическая хирургия является еще одним методом лечения морбидного осложненного ожирения у подростков. Использование фармакотерапии (в комбинации с изменением образа жизни) у детей и подростков с ожирением рекомендуется с 12 летнего возраста при неэффективности мероприятий, направленных на формирование здорового образа жизни, длительность которых составляла не менее 1 года. Единственный препарат, разрешенный для лечения ожирения у детей старше 12 лет в мире и Российской Федерации – это орлистат. Орлистат является ингибитором желудочной и панкреатической липаз, которые участвуют в гидролизе триглицеридов и необходимы для всасывания жиров в тонком кишечнике. В результате действия препарата нарушается расщепление пищевых жиров и уменьшается их всасывание.
Прогноз(благоприятый, если нет осложнеий(СД,АГ…)) и профилактика ожирения. Детям и подросткам с избыточной массой тела и ожирением рекомендуется динамическое наблюдение с контролем антропометрических показателей, оценкой ИМТ и фактического питания
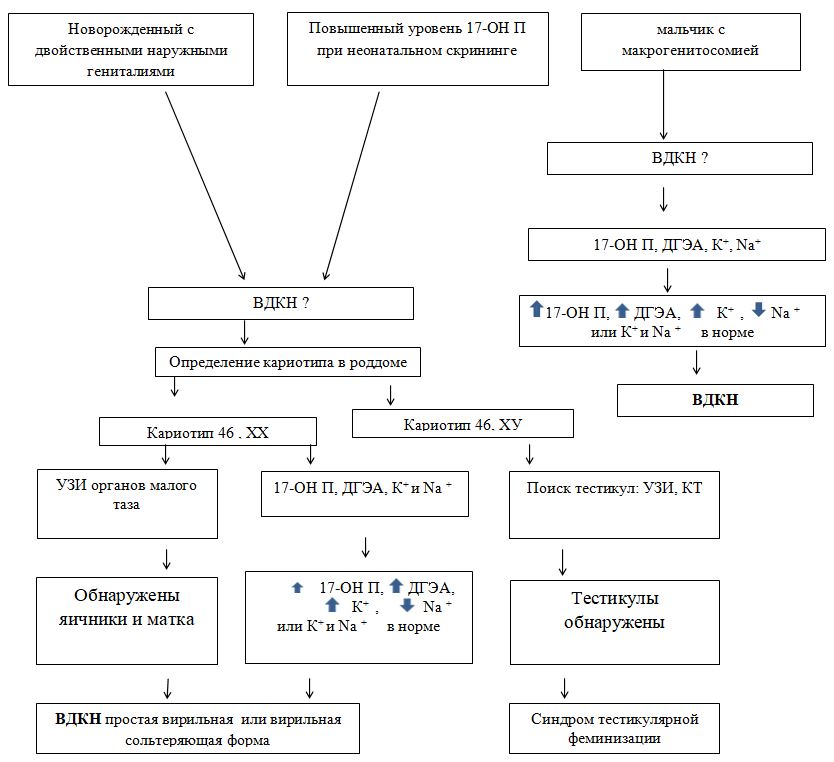
115. Врожденная дисфункция коры надпочечников: определение, эпидемиология, патогенез, классификация, клинические формы (сольтеряющая, вирильная, гипертоническая), клинические особенности сольтеряющей формы у новорожденных и грудных детей, диагностика, ДНК-диагностика, неонатальный скрининг, дифференциальный диагноз, течение и исходы.
Врожденная дисфункция коры надпочечников (адреногенитальный синдром, врожденная надпочечниковая гиперплазия) – группа заболеваний с аутосомно-рецессивным типом наследования, в основе которых лежит дефект одного из ферментов или транспортных белков, принимающих участие в биосинтезе кортизола. Наиболее частое из наследуемых заболеваний.
Патогенез. Причиной развития любой формы ВДКН являются мутации генов, отвечающих за синтез ферментов или транспортных белков, участвующих в биосинтезе кортизола. Снижается секреция кортизола, а для компенсации этого процесса в гипофизе усиливается синтез АКТГ — гормона, вызывающего компенсаторную гиперплазию коры надпочечников для стимуляции выработки кортикостероидов. Классификация:
Сольтеряющий. Самый тяжелый вариант патологии, проявляющийся в первый год жизни ребенка грубыми нарушениями строения наружных половых органов у девочек и их увеличением у мальчиков. Активность 21-гидроксилазы составляет не более 1%. Значительное нарушение стероидогенеза приводит к выраженным соматическим нарушениям — рвоте, поносу, судорогам, чрезмерной пигментации кожи. Без лечения такие дети умирают в раннем возрасте.
Простой вирильный. Течение заболевания менее тяжелое, чем при сольтеряющем варианте. Преобладают проявления неправильного развития гениталий у младенцев женского пола, увеличение их размеров у мальчиков. Признаки надпочечниковой недостаточности отсутствуют. Уровень активности 21-гидроксилазы снижен до 1-5%. С возрастом у пациентов нарастают признаки вирилизации вследствие стимулирующего действия андрогенов.
Неклассический (постпубертатный). Наиболее благоприятная форма АГС, явные признаки которой возникают в период полового созревания и в репродуктивном возрасте. Наружные половые органы имеют нормальное строение, может быть увеличен клитор у женщин и половой член у мужчин. Функциональность 21-гидроксилазы снижена до 20-30%. Заболевание выявляется случайно при обследовании в связи с бесплодием или нарушениями менструальнойфункции.
Сольтеряющий и простой вирильный
При антенатальных формах заболевания основным клиническим симптомом является видимая вирилизация гениталий. У новорожденных девочек обнаруживаются признаки женского псевдогермафродитизма. Клитор большой по размерам или имеет пенисообразную форму, преддверие влагалища углублено, сформирован урогенитальный синус, большие и малые половые губы увеличены, промежность высокая. Внутренние половые органы развиты нормально.
У младенцев-мальчиков увеличен половой член и гиперпигментирована мошонка. Кроме того, при сольтеряющем адреногенитальном расстройстве выражена симптоматика надпочечниковой недостаточности с тяжелыми, зачастую несовместимыми с жизнью соматическими нарушениями (понос, рвота, судороги, обезвоживание и др.), которые проявляются с 2-3-недельного возраста. У девочек с простым вирильным АГС по мере взросления признаки вирилизации усиливаются, формируется диспластическое телосложение.
При сольтеряющей форме имеется дефицит как минералокортикоидов, так и глюкокортикоидов. Причем дефицит последних, при отсутствии компенсации, приводит к развитию смертельно-опасного состояния – сольтеряющего криза, обусловленного снижением реабсорбции натрия в канальцах почек, снижением объем циркулирующей крови, артериального давления, развитием выраженного обезвоживания. Наиболее тяжело сольтеряющие кризы протекают в детском возрасте, с возрастом их частота снижается, однако в стрессовых ситуациях, например, при операциях, травмах, интеркуррентных заболеваниях, они могут осложнить течение заболевания и у взрослых. При вирильной форме отмечается только дефицит кортизола, что при отсутствии лечения проявляется мышечной слабостью, утомляемостью, потемнением кожных покровов на фоне симптомов гиперандрогении. При неклассической форме заболевания ведущими жалобами пациенток являются избыточное оволосение, нарушения менструального цикла, бесплодие или невынашивание беременности.
Диагностика:
Кариотипирование: · обнаружение кариотипа 46,ХХ указывает на генетический женский пол и исключает у ребенка наличие тестис. Биохимический анализ крови: · гиперкалиемия, гипонатриемия, гипохлоремия, повышение активности ренина плазмы (АРП) – при сольтеряющей форме; · гипернатриемия, гипокалиемия, повышение активности АРП – при гипертонической форме. Гормональный профиль* · для ранней диагностики ВДКН – неонатальный скрининг в родильном доме путем определения 17 – ОН прогестерона в сухих пятнах крови новорожденного · у детей и взрослых с ранее недиагностированным заболеванием – определение в крови уровней кортизола, АКТГ, 17 – ОН прогестерона, дигидроэпиандростерона (ДГЭА). При простой вирильной и сольтеряющей вирильной формах наблюдается: сниженный или нормальный уровень кортизола, повышенный уровень АКТГ, 17 – ОН прогестерона, ДГЭА. * Пренатальная диагностика ВДКН не получила широкого клинического применения. Тем не менее доказано, что назначение дексаметазона беременной женщине при положительном результате теста на ВДКН приводит к уменьшению или даже предотвращению вирилизации наружных половых органов у плода. Инструментальные исследования · УЗИ органов малого таза: при кариотипе 46,ХХ обнаруживаются яичники и матка; · КТ надпочечников: двухсторонняя гиперплазия коры; · рентгенография левой кисти: опережение темпов окостенения при простой вирильной и сольтеряющей вирильной формах; · вагинография для уточнения наличия влагалища и его строения.