

304- Органическая химия_Черных В.П. и др_Х., 2007 -776с
.pdfизомериЯ органических соединениЙ
91
Питцера), которое обусловлено взаимодействием (отталкиванием) электронных облаков противостоящих связей. В заслоненной конформации противостоящие связи максимально сближены, поэтому взаимодействие между ними наибольшее. По мере удаления противостоящих связей друг от друга торсионное напряжение уменьшается и становится минимальным в заторможенной конформации. Разность энергий заслоненной и заторможенной конформаций называют энергетическим барьером вращения. Для этана энергетический барьер невелик, он составляет около 12 кДж/моль и легко преодолевается молекулой при обычных температурах за счет энергии теплового движения.
При наличии у атомов, связанных одинарной связью, объемных заместителей (как в молекуле бутана) наряду с торсионным напряжением возникает напря-
жение Ван-дер-Ваальса. Это напряжение обусловлено взаимным отталкиванием заместителей при сближении на расстояние, приблизительно равное сумме их ван- дер-ваальсовых радиусов.
При вращении вокруг связи С-2—С-3 возможны четыре крайние конформации, из которых две заторможенные и две заслоненные:
CH3
H |
H |
H |
H |
CH3
анти-
|
CH3 |
H |
CH3 |
H |
H |
|
H |
|
гош- |
заторможенные конформации
H3C CH3 HCH3
H |
H |
H |
CH3 |
H |
H |
H |
H |
|
|
заслоненные конформации
Заторможенную конформацию, в которой объемные заместители максимально удалены друг от друга (торсионный угол равен 180°), называют антиконформацией. Заторможенную конформацию с торсионным углом между объемными группами,
равным 60°, называют гошконформацией.
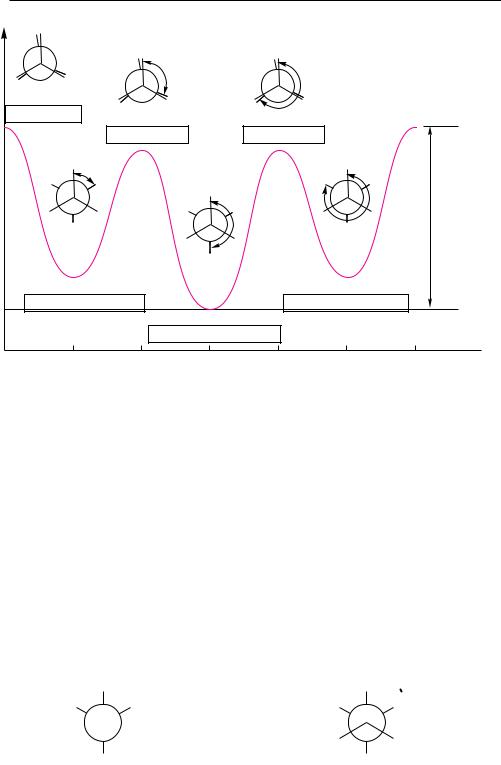
Согласно диаграмме (рис. 5.12) максимальную потенциальную энергию имеет заслоненная конформация (А), в которой метильные группы расположены друг напротив друга. Для этой конформации в потенциальную энергию вносит вклад, помимо торсионного напряжения, также напряжение Ван-дер-Ваальса, обусловленное взаимодействием не связанных друг с другом метильных групп.
По этой причине данная конформация обладает большей энергией, чем заслоненные конформации В и Д, содержащие в заслоненном положении метильную группу и атом водорода. Заторможенные конформации бутана также энергетически неодинаковы. Заторможенные гош-конформации Б и Е за счет метил-метильного взаимодействия обладают несколько большей потенциальной энергией, чем анти-конформация Г, где взаимодействие между объемными группами вообще отсутствует. Поэтому наиболее выгодной для молекулы бутана является анти-конформация.
Хотя вращение вокруг центральной углерод-углеродной связи в бутане связано с преодолением энергетического барьера порядка 25,5 кДж/моль, этот барьер не настолько велик, чтобы препятствовать взаимопревращению конформации при комнатной температуре. Однако в каждый момент времени большая часть молекул представлена наиболее энергетически выгодной конформацией.
92 |
|
|
|
|
|
|
|
|
|
|
|
Глава 5 |
|
|
|
|
|
|
|
|
|
|
|
|
|
Å |
Í3C CÍ3 |
|
|
ÍCÍ3 |
|
|
ÍCÍ3 |
|
|
|
|
|
|
|
|
Í |
|
|
|
|
|
|
|
||
|
Í |
|
|
|
|
|
|
|
|
|
|
|
|
|
Í |
|
|
|
|
|
|
|
|
|
|
|
Í |
À |
|
Í |
CÍ3 |
|
Í |
|
Í |
|
|
|
|
Заcлоненная |
Í |
Í |
|
Í3C |
Ä |
Í |
|
|
|
||
|
|
|
|
|
 |
|
|
|
|
|
|
|
|
|
|
|
Заcлоненная |
|
Заcлоненная |
|
|
|
|||
|
|
Í |
CÍ3 |
|
|
|
|
|
CÍ3 |
|
|
|
|
|
|
CÍ3 |
|
CÍ3 |
|
|
Í3C |
Í |
êÄæ/ìîëü |
|
|
|
|
|
|
|
Í |
|
|
|
|
|
||
|
|
Í |
Í |
Í |
|
Í |
|
Í |
Í |
|
||
|
|
|
|
Í |
|
Í |
|
Í |
|
|
||
|
|
|
|
|
|
|
|
|
25,5 |
|
||
|
|
|
|
|
|
CÍ3 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Á |
|
|
|
|
|
Å |
|
|
|
|
|
Заторможенная (ãîø) |
à |
|
|
Заторможенная (ãîø) |
|
|
||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Заторможенная (àíòè) |
|
|
|
|
|||
|
0 |
|
60 |
|
120 |
180 |
|
240 |
300 |
|
360 |
ϕ, ãðàä |
|
|
|
Рис. 5.12. Диаграмма потенциальной энергии конформаций бутана |
|
||||||||
Конформации с наименьшим запасом энергии называют конформерами или кон формационными (поворотными) изомерами.
Так, бутан при 25 °С существует примерно на 70 % в форме анти-конформера и на 30 % — гош-конформера. С понижением температуры уменьшается энергия теплового движения и, следовательно, повышается содержание более устойчивого конформера, а с повышением температуры, наоборот, увеличивается процентное содержание энергетически менее выгодных конформеров. Конформеры определяют свойства соединения.
Вотличие от конфигурационных изомеров, конформеры превращаются друг
вдруга без разрыва химических связей и не поддаются разделению. Они обнаруживаются только физико-химическими методами.
Следует отметить, что далеко не всегда заторможенная анти-конформация обладает наибольшей устойчивостью. Для ряда органических соединений энергетически более выгодными оказываются заторможенные гош-конформации. Так,
вмолекуле этиленхлоргидрина ClСН2СН2ОН из-за образования внутримолекулярной водородной связи между гидроксильной группой и атомом хлора более стабильной является заторможенная гош-конформация:
OH |
O—H |
H H
H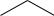 H
H
Cl
H |
Cl |
H |
H |
|
H |
анти-конформация |
гош-конформация |
Глава 6
КИСЛОТНОСТЬ И ОСНОВНОСТЬ ОРГАНИЧЕСКИХ СОЕДИНЕНИЙ
Для оценки кислотности и основности органических соединений в современной органической химии используют две теории — протонную (протолитическую)
теорию Брёнстеда и электронную теорию Льюиса.
6.1. КИСЛОТНОСТЬ И ОСНОВНОСТЬ ПО ТЕОРИИ БРЁНСТЕДА
Согласно теории Брёнстеда кислотой называют любое вещество, способное отдавать протон (донор протона), а основанием — вещество, способное присоединять протон (акцептор протона).
Отсюда теория получила название «протонной» или «протолитической». Для взаимодействия с протоном основание должно иметь неподеленную пару электронов или π-молекулярную орбиталь.
Кислотность и основность являются относительными свойствами вещества. Кислотный характер может проявляться лишь в присутствии основания, и, наоборот, основный характер — только в присутствии кислоты. В целом кислотноосновный процесс состоит в переносе протона от кислоты к основанию и может быть представлен следующей схемой:
А—Н + B |
|
|
|
А– |
+ В+—Н |
|
|
||||
|
|
|
|||
кислота основание |
|
|
|
cопряженное |
сопряженная |
|
|
|
|
основание |
кислота |
Кислота А—Н, отдав протон, превращается в основание А–, которое называют сопряженным основанием данной кислоты. Основание В, присоединив протон,
переходит в сопряженную кислоту В+—Н.
Кислота А—Н и основание А–, а также основание В и кислота В+—Н явля-
ются сопряженными кислотно-основными парами.
Многие органические соединения могут одновременно обладать свойствами и основания, и кислоты. Такие вещества называют амфотерными.
Мерой силы кислоты А—Н является константа кислотности Kа ( a от англ. acid — кислота), которая обычно определяется по отношению к стандартному основанию — воде:
А–Н + Н2О 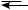
 А– + Н3О+ В сильно разбавленном растворе Ка рассчитывают по формуле
А– + Н3О+ В сильно разбавленном растворе Ка рассчитывают по формуле
Ka = [A–][H3O+ ]. [A − H]
Чем больше значение Kа, тем сильнее кислота. Как правило, константы кислотности очень малы. Например, для уксусной кислоты Kа при 25 °С рав-

Глава 6
94
Йоханнес николаус БрЁнсТед
(1879—1947)
Датский физикохимик. Основные научные работы посвящены химической кинетике, катализу и термодинамике растворов.
Сформулировал (1929) основные положения теории кислот и оснований. Развил (1929) теорию кислотноосновного катализа.
на 1,76·10–5. Оперировать такими малыми числами неудобно, поэтому в практической работе чаще пользуются величинами рKа.
рKа = –lgKa.
Так, рKа уксусной кислоты равна 4,75. Чем меньше значение рKа, тем сильнее кислота.
Подобно кислотам, силу оснований количественно выражают константой основности Кb (от англ. base — основание). Константа основности основания В в воде определяется из равновесия:
В + H2O 
 B+–H + HO–
B+–H + HO–
Kb = [B– − H][HO– ]. [B]
Чем больше Kb, тем сильнее основание. Как и в случае с кислотами, для удобства силу оснований выражают обычно величиной рKb.
рKb = –lgKb.
Чем меньше рKb, тем сильнее соответствующее основание. Однако чаще всего силу оснований оценивают константой кислотности сопряженной основанию кислоты В+—Н, обозначаемую как рKВН+. Чем больше значение рKВН+, тем сильнее основание.
6.1.1. ТиПы органических кисЛоТ
Органические кислоты классифицируют в зависимости от природы кислот-
ного центра (атома элемента, с которым связан атом водорода, обусловливающий кислотные свойства):
онКислоты: карбоновые кислоты, спирты, фенолы, вода и др. SнКислоты: тиолы, тиоловые кислоты и др.
NнКислоты: амины, амиды кислот, имиды и др.
снКислоты: соединения, содержащие сильно полярные С—Н-связи. Известны также SiH-, PH-, AsH-кислоты.
Сила кислот определяется устойчивостью образующихся после отщепления про-
тона сопряженных оснований (анионов). Чем устойчивее сопряженное основание,
тем сильнее кислота. Устойчивость же аниона обусловлена степенью делокализации отрицательного заряда и зависит от ряда факторов:
природы кислотного центра; характера заместителя, связанного с кислотным центром; природы растворителя.
При равных других факторах устойчивость анионов, а следовательно, и кис-
лотность возрастают с увеличением электроотрицательности и поляризуемости
кисЛоТносТь и осноВносТь органических соединениЙ
95
атомов кислотного центра. Поскольку в пределах периода периодической системы электроотрицательность атомов возрастает слева направо (поляризуемость не изменяется), то ОН-кислоты сильнее соответствующих NН-кислот, а те, в свою очередь, сильнее СН-кислот.
CH3—C О |
CH3—C О |
CH3—C О |
O—H |
NH—H |
СH2—H |
уксусная кислота |
ацетамид |
ацетон |
рKа = 4,7 |
рKа = 15,1 |
рKа = 20,0 |
В пределах группы периодической системы электроотрицательность атомов уменьшается сверху вниз, но увеличивается их объем, а следовательно, возрастает поляризуемость, то есть возможность делокализации внешнего электронного облака. Это способствует повышению стабильности аниона и приводит к возрастанию кислотности. Поэтому SН-кислоты обладают большей кислотностью, чем ОН-кислоты:
CH3—CH2—OH |
CH3—CH2—SH |
этанол |
этантиол |
рKа = 18,0 |
рKа = 10,5 |
Органические кислоты с одинаковыми радикалами в зависимости от природы кислотного центра можно расположить по возрастанию кислотности:
СН-кислоты < NН-кислоты < ОН-кислоты < SН-кислоты
Кислотность органических кислот
В пределах отдельного типа кислот кислотность зависит от строения радикала,
связанного с кислотным центром. Алкильные радикалы, благодаря +I-эффекту, увеличивают электронную плотность в кислотном центре и тем самым дестабилизируют анион, что приводит к уменьшению кислотности. Ароматические радикалы, наоборот, повышают устойчивость аниона за счет делокализации отрицательного заряда и способствуют увеличению кислотных свойств.
|
|
|
|
—OH |
|
СH3 OH |
|||||
|
|
||||
метанол |
фенол |
||||
рKа = 16,0 |
рKа = 9,9 |
||||
Заместители, введенные в алифатические и ароматические радикалы, оказывают влияние на кислотность вследствие проявления ими электронных индуктивного и мезомерного эффектов. Электронодонорные заместители (+І; +М-эффект) понижают кислотность, а электроноакцепторные (–І; –М-эффект) — увеличивают ее. Так, введение в молекулу фенола электроноакцепторных групп приводит к повышению кислотности, введение же электронодонорных групп — понижает кислотность.
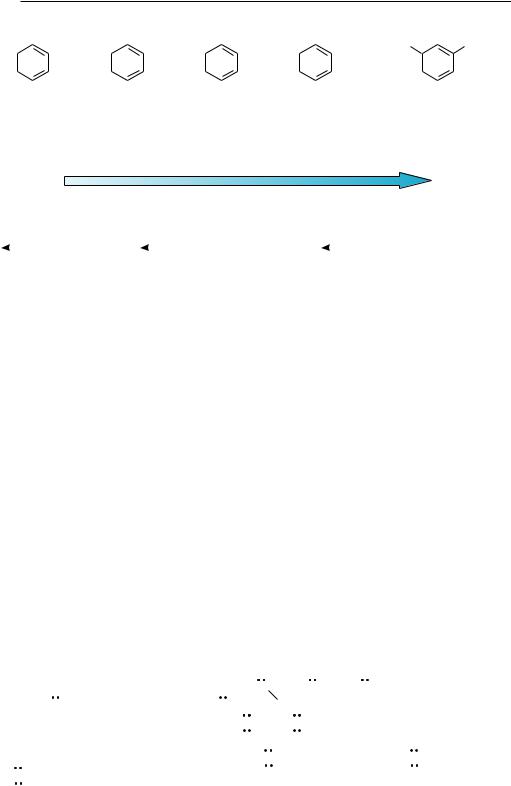
Глава 6
96
|
OH |
|
OH |
|
OH |
|
OH |
|
|
OH |
||||||
|
|
|
|
|
|
|
|
|
|
|
|
O2N |
|
|
|
NO2 |
|
|
|
|
|
|
|
|
|
|
|
|
|
||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
NH2 |
|
СH3 |
|
|
|
|
NO2 |
|
|
NO2 |
|||||
4-аминофенол |
4-метилфенол |
фенол |
4-нитрофенол |
2,4,6,-тринитрофенол |
||||||||||||
pKa = 10,7 |
pKa = 10,1 |
pKa = 9,9 |
pKa = 7,1 |
|
pKa = 0,8 |
|||||||||||
Кислотные свойства фенолов
В алифатическом ряду наиболее сильное влияние на кислотность оказывают заместители, расположенные к кислотному центру ближе.
Cl |
|
CH2—COOH |
Cl |
|
CH2—CH2—COOH |
Cl |
|
CH2—CH2—CH2—COOH |
|
|
|
||||||
хлоруксусная кислота |
β-хлорпропионовая кислота |
|
|
γ-хлормасляная кислота |
||||
|
|
pKa = 2,86 |
|
|
pKa = 4,1 |
|
|
pKa = 4,5 |
Наряду с природой кислотного центра и строением радикала значительное влияние на проявление кислотных свойств оказывает растворитель. Влияние растворителя определяют его диэлектрической проницаемостью и способностью сольватировать растворенные частицы. Чем выше диэлектрическая проницае-
мость растворителя и сольватационный эффект, тем стабильнее ионы в растворе.
При равных прочих условиях сольватация аниона протекает тем сильнее, чем меньше его размер и менее делокализован в нем заряд. Таким образом, влияние сольватационного эффекта растворителя и влияние заместителей на кислотность противоположны друг другу. Наиболее эффективным растворителем является вода, обладающая высокой диэлектрической проницаемостью (ε = 80 при 20 °С) и способностью к сольватации растворенных частиц.
6.1.2. ТиПы органических осноВаниЙ
Как отмечалось ранее, согласно протонной теории в роли основания может выступать любое вещество, способное присоединять протон. Для образования химической связи с протоном основание должно иметь неподеленную пару электронов или π-молекулярную орбиталь.
Органические основания в зависимости от природы основного центра (атом с неподеленной парой электронов или электроны π-связи) подразделяются на n-ос- нования и π-основания.
В n-основаниях центром основности является атом с неподеленной парой электронов. По природе центра основности n-основания классифицируют на следующие типы:
аммониевые (центр основности —N , —N—, —N): амины, азометины (R—CH—N—R), нитрилы (R—C—N), азотсодержащие гетероциклы;
, —N—, —N): амины, азометины (R—CH—N—R), нитрилы (R—C—N), азотсодержащие гетероциклы;
оксониевые (центр основности —O—, —O): спирты, простые эфиры, альдегиды, кетоны, сложные эфиры, амиды кислот и др.;
сульфониевые (центр основности —S—): тиоспирты (R—SH), тиоэфиры
(R—S—R).
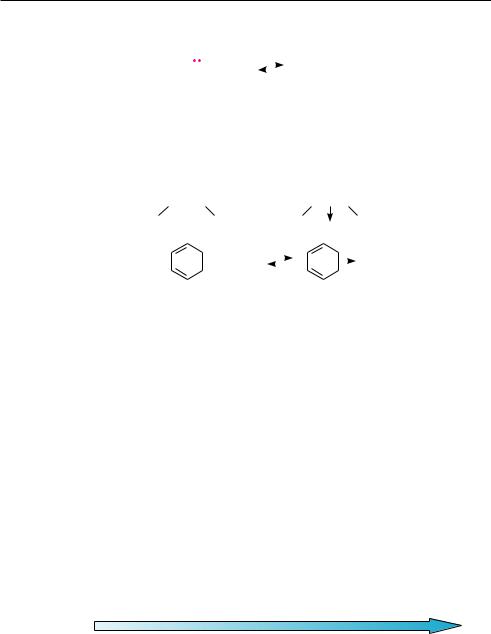
кисЛоТносТь и осноВносТь органических соединениЙ
97
При взаимодействии любого из n-оснований с протоном в качестве сопряженной кислоты образуется соответствующий катион:
|
В + H+ |
|
|
|
B+—H |
|
|
|
|||
|
|
|
|
||
n-основание |
сопряженная кислота |
|
|
|
|
В π-основаниях центром основности являются электроны π-связи. К этой группе оснований относятся алкены, алкадиены, арены. Они являются очень слабыми основаниями по сравнению с n-основаниями. В процессе взаимодейсвия протона с π-основанием происходит частичное перекрывание s-орбитали протона со связывающей π-МО основания, в результате чего образуется так называемый π-комплекс:
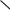 С—С
С—С + H+
+ H+ 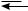
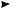
 С—С
С—С
|
|
|
|
|
H+ |
|
|
|||||
π-основания |
|
|
|
|
|
π-комплексы |
||||||
|
|
|
+ H+ |
|
|
|
|
|
|
|
H+ |
|
По аналогии с кислотами сила оснований зависит от ряда факторов:
природы основного центра; характера заместителя, связанного с основным центром; природы растворителя.
При равных других факторах с увеличением электроотрицательности атома основного центра, в пределах одного и того же периода, неподеленная пара электронов удерживается прочнее, а следовательно, основность соединения уменьшается. Поэтому оксониевые основания слабее аммониевых.
В пределах группы периодической системы с увеличением поляризуемости атома основного центра усиливается делокализация неподеленной электронной пары и соответственно уменьшается основность соединения. Поэтому сульфониевые основания слабее оксониевых. Еще более слабыми основными свойствами обладают π-основания, в которых электронная пара, присоединяющая протон, не является свободной.
Таким образом, органические основания в зависимости от природы основного центра можно расположить по возрастанию основности:
π-основания |
< |
сульфониевые |
< |
оксониевые |
< |
аммониевые |
|
|
основания |
|
основания |
|
основания |
|
|
|
|
|
|
|
Основность органических оснований
Большое влияние на основность органических соединений оказывает природа заместителя, связанного с основным центром. Электронодонорные заместители повышают электронную плотность в основном центре и приводят к увеличению основности; электроноакцепторные, наоборот, снижают электронную плотность, а следовательно, уменьшают основность. Например, за счет электронодонорного влияния алкильных групп основность алифатических аминов значительно выше, чем ароматических, где в результате сопряжения неподеленной пары атома азота с π-электронной системой кольца бензольное кольцо проявляет электроноакцепторный характер:

Глава 6
98
CH3 NH2 |
|
δ– |
—NH2 |
||
|
|
|
|
|
|
|
|
|
|
||
метиламин |
|
анилин |
|||
рКBH+ = 10,62 |
рКBH+ = 4,58 |
||||
Влияние растворителя на основность определяется главным образом эффектом сольватации. Как и в случае с кислотами, сольватационный эффект растворителя и электронные эффекты заместителей оказывают на основность противоположное воздействие.
6.2. КИСЛОТЫ И ОСНОВАНИЯ ЛЬюИСА
Американский ученый Г. Н. Льюис в 1923 году предложил электронную теорию кислот и оснований, которая не противоречит теории Брёнстеда, но является более общей. Согласно теории Льюиса основание — любая частица (атом, молекула или анион), способная отдавать электронную пару для образования ковалентной связи, а кислота — любая частица (атом, молекула, катион), способная принимать пару электронов с образованием ковалентной связи.
Основание по теории Льюиса является донором, а кислота — акцептором пары электронов. Из приведенного определения видно, что основания Льюиса тождественны основаниям Брёнстеда. Однако кислоты Льюиса охватывают более широ-
кий круг органических соединений. Кислотой Льюиса считается любая частица, имеющая вакантную орбиталь.
Если в теории Брёнстеда кислота — это донор протона, то согласно теории Льюиса сам протон является кислотой, поскольку имеет вакантную орбиталь. Таким образом, в представлении электронной геории кислота Брёнстеда является соединением, которое образует кислоту Льюиса. Поэтому согласно теории Льюиса к кислотам относят не только соединения, отщепляющие протон (протонные кислоты), но и другие вещества, имеющие вакантную орбиталь и способные принимать пару электронов (апротонные кислоты). Кислотами Льюиса являются такие соеди-
нения, как BF3, AlCl3, SbCl3, ZnCl2, HgCl2 и др.
Кислотно-основный процесс по Льюису состоит в образовании ковалентной связи между основанием и кислотой за счет электронной пары основания и вакантной орбитали кислоты. Так, основания Льюиса, имеющие неподеленные пары электронов, образуют с кислотами Льюиса n-комплексы:
|
|
|
|
|
|
|
|
|
|
|
+ |
– |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
CH |
3 |
|
Cl |
+ |
AlCl |
3 |
|
|
|
CH |
—Cl—AlCl |
3 |
|
CH+[AlCl |
]– |
||
|
|
|
|
|
|||||||||||||
|
|
|
|
|
3 |
|
|
3 |
4 |
|
|||||||
хлорметан |
|
алюминия хлорид |
|
|
|
n-комплекс |
|
|
|
|
|
|
|||||
(основание Льюиса) |
|
(кислота Льюиса) |
|
|
|
|
|
|
|
|
|
|
|
||||

кисЛоТносТь и осноВносТь органических соединениЙ
99
Основания Льюиса, содержащие в своей структуре π-связь, образуют с кислотами Льюиса π-комплексы:
|
+ |
Br+ |
|
|
|
|
|
|
Br+ |
|
|
|
|||||||
|
|
|
|
|
|
|
|||
|
|
|
|
|
|
||||
бензол |
|
катион брома |
|
|
|
π-комплекс |
|||
(основание Льюиса) |
(кислота Льюиса) |
|
|
|
|
|
|||
Легкость протекания кислотно-основной реакции определяется силой кислоты и основания, а также жесткостью или мягкостью кислоты и основания. Представление о жестких и мягких кислотах и основаниях (ЖМКО), введенное Р. Пирсоном, по существу является дальнейшим развитием теории Льюиса. Согласно концепции Пирсона кислоты и основания Льюиса делятся на жесткие
имягкие.
Кжестким кислотам относят кислоты Льюиса, в которых атомы-акцепторы имеют малый объем и несут высокий положительный заряд, а следовательно,
обладают высокой электроотрицательностью и низкой поляризуемостью (H+,
+ + + 2+ 2+ 2+ 3+ 3+ —+—
Li , Na , K , Mg , Ca , Mn , Al , Fe , AlCl3, R C O). Нижняя свободная молекулярная орбиталь (НСМО) в жестких кислотах имеет низкую энергию.
К мягким кислотам относятся кислоты Льюиса, в которых атомы-акцепторы имеют большой объем и несут низкий положительный заряд, а поэтому обладают низкой электроотрицательностью и высокой поляризуемостью (Cu+, Ag+, Hg2+, Pt2+, I2, Br2 и др.). НСМО в мягких кислотах имеет высокую энергию.
К жестким основаниям относят основания Льюиса, в которых атомы-доноры
имеют высокую электроотрицательность и низкую поляризуемость (H2O, OH–,
F–, Cl–, CH3COO–, SO42–, CO32–, NO–3 , R—OH, R—O–, R—O—R, NH3, R—NH2, H2N—NH2, NH2– и др.). Верхняя занятая молекулярная орбиталь (ВЗМО) в жес-
тких основаниях обладает низкой энергией.
К мягким основаниям относят основания Льюиса, в которых атомы-доноры имеют низкую электроотрицательность и высокую поляризуемость (RSH, RS–,
R—S—R, HS–, I–, CN–, R—CN, C2H4, C6H6, H–, R– и др.), ВЗМО в мягких ос-
нованиях обладает высокой энергией.
Исходя из общего положения о том, что более эффективно протекает взаимодействие между орбиталями с близкими энергиями, жесткие кислоты преимущественно реагируют с жесткими основаниями, а мягкие кислоты — с мягкими основаниями (принцип ЖМКО).
Следует отметить, что понятия «жесткие» и «мягкие» кислоты и основания не связаны с понятиями «сильные» и «слабые» кислоты и основания. Так, мягкое основание Н– и жесткое — С2Н5О– — являются сильными, а мягкое основание НS– и жесткое СН3СОО– являются слабыми основаниями.

Глава 7
МЕТОДЫ УСТАНОВЛЕНИЯ СТРОЕНИЯ ОРГАНИЧЕСКИХ СОЕДИНЕНИЙ
Прежде чем приступить к изучению строения органического соединения, химик должен выделить из реакционной смеси или природных источников индивидуальное вещество в чистом виде и провести оценку его чистоты.
Для выделения и очистки органических соединений обычно используют перекристаллизацию, перегонку, экстракцию, хроматографию и другие методы. Для оценки чистоты определяют физические константы (температура плавления или кипения), хроматографические характеристики, показатель преломления и др. Подробное описание методов выделения, очистки и доказательства индивидуальности органических соединений приведено в практических руководствах по органической химии.
После получения вещества в химически чистом виде устанавливают его строение (структуру), то есть определяют природу и количество атомов, входящих в состав молекулы, последовательность их связывания, расположение в пространстве и тип химической связи между ними.
Существует два основных подхода к установлению строения органических соединений. Если исследуемое вещество было ранее изучено, для доказательства его структуры определяют физические константы и спектральные характеристики, которые сравнивают с литературными данными. Если же органическое соединение получено впервые, его сначала подвергают качественному и количественному элементному анализу, то есть устанавливают, какие элементы
ив каком количестве входят в состав (см. практические руководства по органической химии). Затем определяют молекулярную массу вещества. Для этой цели применяют криоскопический (по понижению температуры замерзания)
иэбулиоскопический (по повышению температуры кипения) методы, которые рассматриваются в курсе физической химии. В настоящее время для определения молекулярной массы широко используют метод масс-спектрометрии.
На основании молекулярной массы и данных элементного анализа устанавливают молекулярную формулу (брутто-формулу) вещества. Наконец, определяют структуру углеродного скелета, природу и положение функциональных групп,
устанавливают определенные фрагменты молекулы и расположение атомов в пространстве. Для этих целей используют химические и физические (инструментальные) методы. На основании полученных данных выводят структурную или стереохимическую формулу.
7.1. ХИМИЧЕСКИЕ МЕТОДЫ
Применение химических методов для установления строения органических соединений основано на использовании качественных реакций, позволяющих