

Физ химия и физ т.д
..pdfХ и м и ч е с к а я к и н е т и к а
На известную неопределенность понятия индукционного периода мы уже указывали при рассмотрении последовательных реакций (см. 2.2.1.5.А). Фор-
мально его можно определить как время, в течение которого скорость реакции возрастает в e раз; экспериментальное определение продолжительности τинд фактически зависит от чувствительности прибора.
Следует отметить, что на рис. 2.16 обозначен еще и третий предел воспламенения, лежащий при более высоких давлениях. По-видимому, в большинстве случаев его существование связано с развитием теплового взрыва, хотя при некоторых обстоятельствах в условиях высокого давления не исключается и цепное воспламенение.
Из двух вариантов теории цепных реакций – строгого, основанного на решении системы дифференциальных уравнений, и менее строго вероятностного варианта, мы остановимся на последнем, являющемся несравненно более наглядным.
Нами ранее уже было выведено понятие средней длины цепи L, являющейся средним числом звеньев цепи, вызываемым одной активной частицей (атомом или радикалом), возникшей первоначально каким–либо независимым путем – фотохимическим или другим. Пусть число таких независимо возникающих в единицу времени в единице объема частиц равно n0 . Очевидно n0 можно
назвать скоростью зарождения цепей.
Скорость цепных реакций зависит от длины цепи, которая в свою очередь определяется соотношением скоростей обрыва и продолжения цепи. Обрыв цепи в одном из ее звеньев характеризуется вероятностью обрыва цепи β – ве-
личиной обратной средней длине цепи. Тогда вероятность продолжения цепи будет равна (1 − β ).
Для цепной неразветвленной реакции длина цепи равна |
|
L = (1 − β )/ β , |
(2.179) |
т.е. длина цепи равна отношению вероятностей продолжения и обрыва цепи. Если вероятность обрыва цепи мала (β 1),то вместо уравнения (2.179)
можно записать |
|
L =1/ β . |
(2.180) |
Следовательно, вероятность обрыва цепи |
|
β =1/ L . |
(2.181) |
Собственно скорость неразветвленной цепной реакции равна произведению скорости зарождения цепи на длину цепи:
v = n0 L . |
(2.182) |
71

Х и м и ч е с к а я к и н е т и к а
Отсюда следует, что на каждый акт зарождения цепи приходится L звеньев цепной реакции.
Скорость неразветвленной цепной реакции определяется течением всех ее трех стадий, поскольку зависит от скорости зарождения цепи и длины цепи. Последняя связана с условиями продолжения и обрыва цепи.
Допустим возможность разветвления цепи с некоторой вероятностью δ . Введем еще некоторые определения и обозначения. Пусть τз – время, в течение
которого в среднем протекает одно звено цепи. Тогда произведение Lτз будет
определять среднее время прохождения всей цепи от момента зарождения до обрыва. Обозначим через n концентрацию активных частиц, т.е. их число в единице объема. Скорость изменения концентрации частиц выразится разностью скорости их образования и исчезновения. Подробнее остановимся на последней. В том случае, когда длина цепи L =1, т.е. собственно цепи отсутствуют, частица гибнет в каждом звене, и за время τз прореагируют все n наличных
частиц, а скорость исчезновения будет равна n /τз. Если же цепи развиваются и
их средняя длина равна L, то частица в среднем будет регенерироваться L раз и гибнуть только через время Lτз . В этом случае уменьшение концентрации час-
тиц выразиться, с учетом (2.181), соотношением
n = β n .
Lτз τз
Далее учтем возможность разветвления, т.е. пусть δ > 0 .Приближенно влияние разветвления можно учесть, считая, что оно действует как бы в направлении, обратном обрыву, т.е. удлиняя цепи и уменьшая вероятность обрыва до (β −δ ). Тогда скорость изменения концентрации активных частиц можно
окончательно записать так:
dn |
= n |
− (β −δ ) |
n |
. |
(2.183) |
dτ |
|
||||
0 |
τз |
|
|||
Разделяя переменные и интегрируя при условии, что в начале реакции τ =0 и n = 0 , получаем зависимость n от времени:
|
τ |
n |
|
|
− |
β−δ |
τ |
|
|
|
τ |
|
|||||
n = |
|
з 0 |
1 |
− e |
|
з . |
(2.184) |
|
|
|
|||||||
|
β −δ |
|
|
|
|
|
||
|
|
|
|
|
|
|
|
|
С учетом (2.184) можно определить скорость реакции, т.е., скажем, скорость
прироста концентрации молекул продукта. Поскольку в одном звене, т.е. за время τз , появляется одна молекула, то общее число молекул, образующихся в
единице объема за единицу времени, будет n /τ . Следовательно, скорость реакции выразится соотношением
72
Х и м и ч е с к а я к и н е т и к а
|
n |
|
n |
|
|
− |
β−δ |
τ |
|
|
|
|
|
τ |
|
|
|||||
v = |
|
= |
0 |
|
1 −e |
|
з |
, |
(2.185) |
|
τз |
β − |
|
||||||||
|
|
δ |
|
|
|
|
|
|||
|
|
|
|
|
|
|
|
|
|
|
которое и является основным уравнением излагаемого варианта цепной теории. При анализе уравнения (2.185) следует рассмотреть несколько случаев.
Простейший случай – отсутствие разветвлений, т.е. уравнение (2.185) при этом принимает вид
|
|
|
− |
β |
τ |
v = Ln |
1 |
−e |
|
τз . |
|
0 |
|
|
|
|
|
|
|
|
|
|
|
δ =0 . С учетом (2.180)
(2.186)
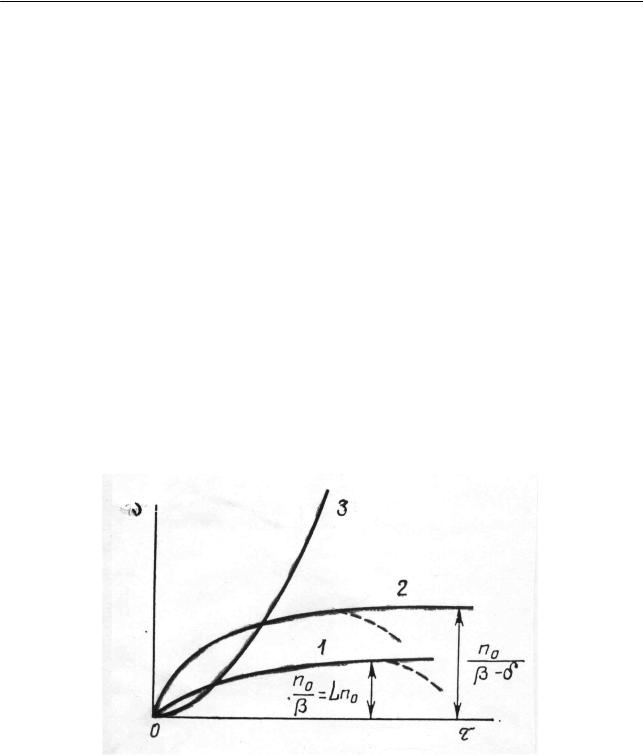
Как показано на рис. 2.20, кривая 1, скорость реакции с момента ее начала должна увеличиться и достичь предела, равного Ln0 = n0 / β , что соответствует
раннее полученному выражению (2.182) для скорости неразветвленной цепной
реакции. Таким образом, в рассматриваемом случае система должна достигать стационарного состояния, в котором скорость постоянная. Вследствие наличия цепей эта стационарная скорость в L раз больше скорости зарождения цепей n0 ,
т.е. скорости реакции в отсутствие цепей (L =1).
Рис. 2.20. Зависимость скорости цепной реакции от времени:
1 – отсутствие разветвления, 2 – вероятность разветвления меньше вероятности обрыва (0 <δ < β ), 3 – вероятность разветвления больше вероятности обрыва (δ < β )
73

Х и м и ч е с к а я к и н е т и к а
Рассмотрим случай, когда имеется конечная вероятность разветвления, но ее численное значение меньше вероятности обрыва, т.е. 0 <δ < β . Такое соот-
ношение вероятностей также ведет к достижению стационарной скорости (рис. 2.20, кривая 2), но большей, чем в первом случае, а именно n0 / (β −δ ).
Следует иметь ввиду, что все приведенные рассуждения относятся к идеализированным условиям развития реакции – концентрации исходных веществ поддерживаются постоянными, а продукты реакции выводятся из реакционной зоны. На самом деле при проведении реакции, например, в замкнутом сосуде скорость обязательно пройдет через максимум вследствие «выгорания» исходных веществ (пунктирные линии на рис. 2.20). Может даже случиться, что стационарное значение не будет достигнуто: максимальная скорость окажется меньше стационарной.
В случае, если вероятность разветвления окажется больше вероятности обрыва, т.е. δ > β , наблюдается совсем иная картина. Уравнение (2.185) прини-
мает вид
|
n |
|
|
δτ−βτ |
|
ϕτ |
ϕτ |
|
|
v = |
0 |
|
|
з |
|
= A(e |
−1)≈ Ae |
, |
(2.187) |
δ − |
β e |
|
−1 |
||||||
|
|
|
|
|
|
|
|
|
|
где A и ϕ – положительные |
постоянные. Выражение |
(2.187) |
совпадает с |
||||||
(2.178). Наличие в нем положительной степенной функции времени свидетель-
ствует о возможности беспредельного ускорения реакции (см. рис.2.20, кривая 3, идентичный рис. 2.19). Как уже говорилось, не следует искать несоответствия с реальным процессом формального стремления скорости к бесконечности в выражениях (2.178) и (2.187).
И, наконец, подчеркнем, что в пределах полуострова воспламенения как раз и реализуются условия, когда δ > β , а вне его условия соответствуют неравен-
ству β >δ . Таким образом, теория разветвленных реакций количественно объясняет существование нижнего и верхнего пределов воспламенения.
Условие (2.175) протекания реакции между двумя исходными веществами
как сложной реакции с участием активных промежуточных частиц было сформулировано нами на примере рассмотрение представленной в общем виде реакции (2.173), протекающей по двустадийному механизму. Однако такие про-
стые двустадийные механизмы не типичны для образования активных промежуточных частиц, так как это может быть только при наличии у одного из компонентов реакции некоторых специфических особенностей строения, благоприятствующих превращению в активные частицы. Примером может служить енольная форма ацетона, играющая роль активной частицы в ходе реакции иодирования ацетона.
74

Х и м и ч е с к а я к и н е т и к а
Широкие возможности получения активных промежуточных частиц открываются при использовании дополнительных специально подобранных компонентов. Два основных пути использования дополнительных компонентов основаны на двух фундаментальных явлениях химической кинетики – катализе и индукции.
Весьма детальное самостоятельное рассмотрение явления катализа будет проведено нами несколько позднее (см. 2.4). Здесь же, до того, как перейти к рассмотрению химической индукции, кратко остановимся на кинетике автокаталитических реакций.
Автокаталитические реакции – это реакции, самоускоряющиеся одним из продуктов реакции, являющимся катализатором. Они не являются ката-
литическими в прямом смысле. Дело в том, что в них катализатор, образуясь в качестве конечного продукта, накапливается. Следовательно, концентрация катализатора не остается постоянной – она увеличивается, т.е. не выполняется один из основных признаков катализа, а именно: постоянство концентрации катализатора, ее неизменность к концу каталитического акта.
Гомогенную элементарную автокаталитическую реакцию в общем виде можно записать как
A + B + (D)→ D + E ,
где D – продукт реакции (катализатор).
Зависимость скорости автокаталитической реакции от концентрации, времени и других факторов, а также кинетические уравнения для реагентов имеют различный вид для разных механизмов протекания ее по стадиям, а также от условий ее проведения. Но общим для закрытых систем является появление
максимума на зависимости скорости реакции от концентрации катализатора или времени и наличие индукционного периода, если начальная концентрация продукта-катализатора мала.
Наличие различных форм уравнения для скорости автокаталитических реакций делает целесообразным рассмотрение основных из них на примере конкретных химических реакций, при изучении которых они были получены.
Н.А. Меншуткин обнаружил начальное ускорение образования ацетанилида при разложении третичного амилацетата. На графике в координатах «количество разложенного эфира –– время» была получена S-образная кривая с точкой перегиба при 50% разложения эфира. Д.П. Коновалов доказал, что ускоряющим действием реакция Меншуткина обязана образующейся уксусной кислоте, и дал уравнение скорости в следующем виде:
v = |
dx |
= k (100 |
|
1 |
|
, |
(2.188) |
dτ |
− x) x + 2 |
6 |
P |
||||
|
|
|
|
|
|
75

Х и м и ч е с к а я к и н е т и к а
где x – количество разложившегося эфира в процентах, P – начальная концен-
трация уксусной кислоты, 2 16 – отношение молекулярных масс эфира и кисло-
ты.
Оствальд еще до Коновалова дал уравнение скорости гидролиза уксуснометилового эфира в присутствии заранее добавленной уксусной кислоты, являющейся катализатором:
dx = k (c |
+ x)(c |
− x), |
(2.189) |
|
dτ |
01 |
02 |
|
|
|
|
|
|
|
где c01 – начальная концентрация эфира, x – его прореагировавшее количество, c02 – начальная концентрация уксусной кислоты.
Для дальнейшего рассмотрения запишем уравнение автокаталитической реакции в несколько ином виде, взяв за основу гидролиз уксусноэтилового эфира:
CH3COOC2 H5 + H2O →CH3COOH + C2 H5OH ,
что, с учетом вышеприведенной записи гомогенной автокаталитической реакции в общем виде, идентично следующему:
CH3COOC2 H5 + H2O + H + →CH3COO− + C2 H5OH + 2H + .
Следует отметить, что процессы гидролитического расщепления (омыления) сложных эфиров являются типичными автокаталитическими реакциями. Роль катализатора в них выполняет кислота, а вернее ионы водорода, что наглядно видно из второго варианта записи химической реакции, отражающего механизм ее протекания. В присутствии заранее добавленной сильной кислоты реакция подчиняется обычному уравнению первого порядка с константой скорости, пропорциональной концентрации кислоты. Обозначим, как и в формуле Оствальда, через c01 начальную концентрацию эфира, через x – его прореагиро-
вавшее количество и, следовательно, количество образовавшейся уксусной кислоты. Допустим, что и в первоначальном нейтральном растворе реакция имеет малую, но конечную скорость1 – соответствующая константа скорости равна k0 . Общее уравнение скорости запишем в следующем виде:
v = dx k |
|
(c |
− x)+ kx(c |
− x). |
(2.190) |
dτ |
0 |
01 |
01 |
|
|
В уравнении (2.190) первый член справа соответствует скорости некаталити-
ческой реакции, а второй – скорости каталитической реакции, пропорциональной концентрации катализатора x. Вынося за скобку сначала c01 − x , а затем k,
получаем
1 Можно, например, допустить начальное влияние ионов водорода и гидроксид-ионов, всегда присутствующих в водном растворе.
76

Х и м и ч е с к а я к и н е т и к а
v = dx = k |
|
k0 |
+ x |
|
(c |
− x). |
||||||
|
|
|||||||||||
|
|
dτ |
|
|
|
k |
|
|
01 |
|||
|
|
|
|
|
|
|
|
|
|
|
||
Или, разделяя переменные |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
dx |
|
|
|
|
= kdτ |
||||
|
|
k0 |
|
|
|
|
|
|
|
|
||
|
+ x |
(c |
|
− x) |
|
|||||||
|
|
|
|
|||||||||
|
|
k |
|
01 |
|
|
|
|
|
|||
|
|
|
|
|
|
|
|
|
|
|
|
|
(2.191)
(2.192)
и интегрируя, получаем следующую зависимость количества прореагировавшего вещества от времени:
|
|
|
|
|
( 0 |
|
01) |
|
|
|
||
|
|
k0 e |
k |
+kc |
τ |
−1 |
|
|
|
|||
|
x = c |
|
|
|
|
. |
(2.193) |
|||||
|
|
|
|
|
|
01) |
||||||
|
01 |
kc |
|
+ k e( 0 |
|
|
|
|||||
|
|
01 |
|
|
0 |
k |
+kc τ |
|
|
|||
|
|
|
|
|
|
|
|
|
|
|||
Из уравнения (2.193) видно, что при τ =0 |
x =0 , а при τ →∞ можно пренеб- |
|||||||||||
речь kc |
и 1 по сравнению с бесконечно возрастающими значениями |
k |
+kc τ |
|||||||||
e( 0 |
01) |
|||||||||||
01 |
|
x = f (τ ) |
|
|
|
|
||||||
и тогда |
x = c01 . В целом же, кривая |
– типичная для автокаталитиче- |
||||||||||
ских реакций S-образная кривая с точкой перегиба. Приравнивая вторую производную x по времени нулю, находим время, соответствующее точке перегиба:
|
ln |
c01k |
|
|
|
|||
k0 |
(2.194) |
|||||||
|
|
|
|
|||||
τпер = |
|
|
|
. |
||||
k |
0 |
+ c k |
||||||
|
|
01 |
|
|
|
|||
Как видно из выражения (2.194), это время определяется значениями констант
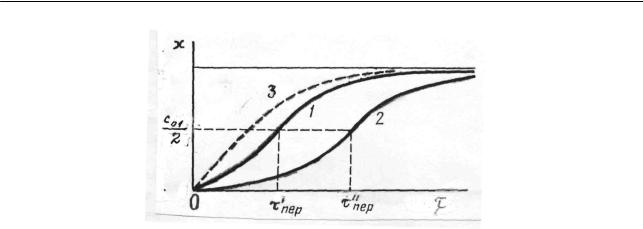
скоростей и при увеличении константы скорости каталитической реакции уменьшается. Отмеченное наглядно видно из рис. 2.21, на котором приведены две кривые зависимости x от времени для различных значений k. Что же касается количества прореагировавшего вещества, соответствующего точке перегиба, то оно практически не зависит от значений констант и равно половине исходного, что можно подтвердить двояко. С одной стороны, подставляя τпер из
(2.194) в исходную формулу для x (2.193) или, с другой стороны, проще – из условия dv / dx = 0 , где v отвечает выражению (2.191). Последнее корректно с
учетом двух вышеотмеченных моментов: 1. x – как количество прореагировавшего эфира, так и количество образовавшейся уксусной кислоты, являющейся катализатором; 2. наличие максимума на зависимости v = f (x). В обоих случа-
ях находим
x = |
с01 |
− |
k0 |
≈ |
c01 |
. |
(2.195) |
|
|
|
|||||
пер |
2 |
|
2k |
2 |
|
|
|
|
|
|
|
||||
77
Х и м и ч е с к а я к и н е т и к а
Рис. 2.21. Зависимости количества прореагировавшего вещества от време-
ни:
1 и 2 – для автокаталитических реакций, у которых константа скорости больше для первой реакции; 3 – для реакции первого порядка
По условию константа k0 мала по сравнению с k , что позволяет пренебречь членом k0 / 2k . Зависимости, приведенные на рис. 2.21, наглядно подтвержда-
ют, что точка перегиба на кривой расходования вещества соответствует, очевидно, наибольшей скорости реакции – угловой коэффициент dx / dτ имеет в этот момент максимальное значение. В связи с этим отметим, что прохождение скорости через максимум при x ≈c01 / 2 вообще характерно для автокаталити-
ческих процессов.
На рис. 2.21 для сравнения приведена и кривая 3, соответствующая обычной некаталитической реакции первого порядка. Для этой кинетической кривой характерен монотонный ход с постоянно убывающим наклоном, т.е. уменьшающаяся с начального момента скорость реакции (см. 2.2.1.1). При малой начальной скорости каталитической реакции, например в случае, соответствующем кривой 2 на рис. 2.21, реакция какое-то время может быть практически вообще незаметна. Другими словами, она идет с индукционным периодом, длительность которого, как уже неоднократно отмечалось, связана с точностью применяемой методики анализа.
Дифференцирование выражения (2.193) по времени позволяет найти зависимость скорости от времени
|
|
|
k |
|
|
1 |
|
|
|
|
|
|
|
|
k |
+kc τ |
|
|
|
|
|
|
|
|
+ |
|
|
|
|
|
(k |
|
+ kc |
)e( 0 |
01) |
|
|
||
|
|
|
|
|
|
|
|
0 |
|
|
|||||||||
|
dx |
|
|
|
c01 |
|
|
|
|
01 |
|
|
|
|
|||||
v = |
= |
k0 |
|
|
|
|
|
|
|
|
|
|
|
. |
(1.196) |
||||
dτ |
|
|
|
k |
|
|
1 |
k |
+kc |
τ 2 |
|||||||||
|
|
|
|
|
|
|
|
||||||||||||
|
|
|
|
|
|
|
|
|
+ |
|
|
|
e( 0 |
01) |
|
|
|
||
|
|
|
|
|
|
|
|
|
c01 |
|
|
|
|||||||
|
|
|
|
|
k0 |
|
|
|
|
|
|
|
|
|
|||||
78

Х и м и ч е с к а я к и н е т и к а
Учитывая, что kc01 / k0 1, можем для малых τ пренебречь в знаменателе экс-
понентой, близкой к единице. Тогда, вводя обозначения для постоянных величин
|
k |
+ |
1 |
|
|
(k |
|
+ kc |
) |
|
|||
|
|
|
|
|
|||||||||
|
|
c |
|
|
|
||||||||
k |
0 |
|
01 |
|
|
|
0 |
01 |
|
|
|||
|
|
|
|
|
|
|
|
|
|
= A |
|||
|
|
|
|
|
|
k |
|
2 |
|
|
|||
|
|
|
|
|
|
|
|
|
|||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||
|
|
|
|
|
k0 |
|
|
|
|
|
|||
и
k0 + kc01 =ϕ ,
получим для начального периода следующую зависимость скорости реакции от времени:
v = Aeϕτ . |
(2.197) |
Это выражение близко к выражению (2.187), выведенному для скорости раз-
ветвленной цепной реакции также без учета расходования исходного вещества. Кинетика автокаталитических и цепных разветвленных реакций бывает весьма сходна. Эти реакции не всегда легко различить при исследовании, хотя в общем природа их существенно различна.
Помимо рассмотренных нами гомогенных, известны также гетерогенные каталитические реакции, катализируемые конечными продуктами. Примерами таких реакций являются, например, растворение меди и ртути в азотной кисло-
те, катализируемое ионами NO2− и оксидами азота; катализируемое образую-
щимся металлом восстановление некоторых твердых оксидов металлов водородом или оксидами азота; взаимодействие твердого оксида серебра с CO2 , кото-
рое катализируется образующимся Ag2CO3 .
И, наконец, отметим, что при реакциях в конденсированных системах, например при разложении взрывчатых веществ, автокаталитическое ускорение процесса сопровождается ускорением за счет разогрева.
Сопряженные реакции – это две реакции, одна из которых индуцирует
протекание другой. Сопряженными реакциями много занимался русский ученый Н.Шилов, воззрениям которого мы будем преимущественно придерживаться при рассмотрении данного вопроса. Термин сопряженные реакции введен Оствальдом, а само явление открыто, по-видимому, Кесслером. Явление, на котором основаны сопряженные реакции (процессы) носит название химиче-
ской индукции. Химическая индукция – это явление, при котором самопро-
извольно идущая в системе реакция вызывает протекание другой реакции, не осуществимой в отсутствии первой (Н.Шилов). Как правило, в явлении
79

Х и м и ч е с к а я к и н е т и к а
можно отличать первичный, самопроизвольный «индуцирующий» процесс от вторичного, индуцируемого, в отсутствии первой реакции не идущего.
Допустим, прямое взаимодействие вещества |
A1 с веществом A2 не осуще- |
|||
ствимо. В то же время, некоторое вещество X может вступать в реакцию с A1 , |
||||
переводя его в активное производное A* |
, реагирующее с веществом A с обра- |
|||
|
1 |
|
|
2 |
зованием продуктов реакции (B1 и B2 ). Само вещество X при этом превращает- |
||||
ся практически необратимо в какой-то продукт Y, т.е. расходуется. Таким обра- |
||||
зом, взаимодействие между веществами |
A1 |
и A2 |
происходит с помощью сле- |
|
дующих элементарных стадий: |
|
|
|
|
X + A → A* |
+Y , |
(2.198) |
||
|
1 |
1 |
|
|
A* + A → B + B . |
(2.199) |
|||
1 |
2 |
1 |
2 |
|
В общем случае можно сказать, что с помощью элементарных стадий (2.198) и (2.199) осуществляется два химических превращения, т.е. протекают
сопряженные реакции
A1 + A2 → B1 + B2 , |
(2.200) |
X →Y . |
(2.201) |
В качестве примера сопряженных реакций рассмотрим окисление бензола в фенол при низкой температуре пероксидом водорода. Стехиометрическое уравнение реакции
C6 H6 + H2O2 →C6 H5OH + H2O. |
(2.202) |
Эта реакция должна идти с синхронным разрывом двух существующих и образованием двух новых связей в четырехчленном циклическом активированном комплексе (вопросы, связанные с активированными комплексами будут рассмотрены в 2.3.2)
что связано с преодолением высокого энергетического барьера. Поэтому прямое взаимодействие компонентов практически не приводит к образованию продуктов реакции (2.202). Если же в исходную смесь добавить соль железа (II),
то в результате реакции
Fe2+ + H2O2 →[FeOH ]2+ + OH
80