

Физ химия и физ т.д
..pdfХ и м и ч е с к а я к и н е т и к а
Рис. 2.41. Кинетические кривые расходования исходного вещества в присутствии катализатора (1), без него (2) и в присутствии ингибитора (3) ( ср – равновесная концентра-
ция)
Таким образом, в отличие от реакций с нетермической активацией реаги-
рующих веществ (см. 2.2.1.5.D), в результате катализа лишь ускоряется
процесс достижения равновесных концентраций исходных веществ и продуктов реакции, но не обеспечивается получение сверхравновесных концентраций (сверхравновесного выхода) продуктов реакции. Поэтому для каталитических реакций термодинамический подход всегда оправдан. В отличие от химической индукции (см. 2.2.1.5.С), катализ не может вызвать про-
текания реакций, для которых в данных условиях ∆G > 0 , а может лишь увеличить скорость реакции в том случае, если ∆G < 0 . В состоянии рав-
новесия (∆G = 0) при катализе имеет место в равной степени ускорение
как прямой, так и обратной реакций. Другими словами, катализатор может одновременно ускорять как прямую, так и обратную реакции, но при этом кон-
станта равновесия см. (2.80) остается постоянной. Иначе говоря, катализатор
не может изменить равновесие термодинамически невыгодных обратимых реакций, у которых равновесие сдвинуто в сторону исходных веществ.
Многочисленные наблюдения привели к двум важным обобщениям в области катализа. Во-первых, уже отмечавшееся снижение энергии активации (рис. 2.40) как наиболее отчетливое проявление активности катализатора. Вовторых, специально для гомогенного катализа в растворах положение о пропорциональности между скоростью и концентрацией катализатора. Указанное позволяет заключить, что катализатор своим веществом участвует в реакции, образуя некоторые неустойчивые промежуточные соединения или комплексы и, таким образом, дает реакции возможность идти по иному более энергетически выгодному пути.
Катализ может быть как гомогенным, так и гетерогенным. При гомогенном катализе катализатор и реагирующие вещества находятся в одной фазе – жидкой или газовой, а при гетерогенном – в разных фазах. Во втором случае хими-
171

Х и м и ч е с к а я к и н е т и к а
ческая реакция протекает на границе раздела фаз. При гетерогенном катализе наиболее часто используется твердый катализатор, а реагирующие вещества находятся в газовой или жидкой фазе. Реакция осуществляется на поверхности твердого катализатора.
Примером гомогенного катализа может служить ускорение гидролиза углеводов в водном растворе в присутствии кислот и окисление CO до CO2 в газо-
вой среде в присутствии паров воды. Примером гетерогенного катализа служит окисление аммиака на поверхности платины.
Возможен и гетерогенно-гомогенный катализ, когда реакция начинается на поверхности твердого катализатора, а продолжается в объеме реакционной среды. Так, при окислении углеводородов RH на поверхности твердого катали-
затора образуются активные частицы из свободных радикалов HO2 и RO2 , ко-
торые затем переходят в газовую фазу и продолжают там цепную реакцию. Среди различных видов катализа наибольшее распространение имеют ки-
слотно-основный, окислительно-восстановительный и ферментативный.
Последний относят к гомогенному катализу. Роль катализаторов здесь выполняют ферменты или энзимы – биокатализаторы, под действием которых в живых организмах протекает синтез большинства веществ, необходимых для их жизнедеятельности. Особое значение ферментов для понимания сущности каталитического действия обусловливает самостоятельное рассмотрение ферментативного катализа (см. 2.3.6).
В случае кислотно-основного катализа катализаторами служат вещества, способные передавать или принимать протон (H + ) или имеющие акцептор
электронной пары, например Al.
К типичным кислотно-основным катализаторам относятся протонные кислоты (H2 SO4 , CH3COOH , HF ), фосфаты, сульфаты, алюмосиликаты. Кислот-
но-основный катализ лежит в основе крекинга, процессов гидратации и дегидратации, гидролиза, некоторых реакций полимеризации и изомеризации.
В зависимости от природы частиц, выступающих в качестве катализатора, все каталитические реакции кислотно-основного типа можно подразделить на четыре группы: 1. специфический кислотный катализ, включающий реакции,
катализируемые только ионами водорода [ионом гидроксония H3O+ или лиония SH + (например, C2 H5OH2+ ), где S – молекула растворителя]; 2. специфиче-
ский основный катализ, включающий реакции, катализируемые гидроксидионами; 3 общий кислотно-основный катализ, включающий реакции, катализируемые любыми кислотами или основаниями Бренстеда, т.е. веществами способными соответственно отдавать или принимать протон; 4 электрофильнонуклеофильный катализ, включающий реакции, катализируемые акцепторами (электрофилами) или донорами (нуклеофилами) электронных пар, в том числе
172

Х и м и ч е с к а я к и н е т и к а
и апротонными кислотами Льюиса, типичными представителями которых яв-
ляются AlBr3 , AlCl3 , FeCl3 , SbCl3 , BF3 , Al (CH3 )Br2 .
При окислительно-восстановительном катализе происходит ускорение реакций, в которых изменяется степень окисления атомов, входящих в молекулы реагирующих веществ. Катализаторами в этих реакциях выступают ионы переходных металлов, например, комплексные ионы меди (I, II ), железа
(II, III ), кобальта (II, I ), молибдена (IV , V , VI ), служащие эффективными по-
средниками реакций с переносом электронов. При этом переход электронов от реагентов к катализатору осуществляется легче, чем от восстановителя к окислителю.
В силу того, что в реакции иона с молекулой при переносе электрона всегда возникает свободный радикал, такие окислительно-восстановителтьные системы являются генераторами свободных радикалов. Они используются для инициирования реакций радикальной полимеризации, окисления, хлорирования. Помимо этого, к окислительно-восстановительным каталитическим реакциям относятся реакции восстановления, гидрирования, дегидрирования и карбоксилирования, а также разложения некоторых кислородсодержащих соединений. Исключительно важную роль оксилительно-восстановительные реакции играют в живом организме, где они составляют основу ферментативных процессов дыхания, фиксации азота и удаления вредных для организма продуктов.
Значительная часть применяемых в промышленности катализаторов была подобрана эмпирически, при этом были сформулированы основные требова-
ния к катализатору: он должен обладать каталитической активностью, быть специфичным, селективным, а также механически прочным, термостойким и способным к регенерации.
Каталитическая активность или просто активность, характеризует способность катализатора ускорять химическую реакцию. Количественно каталитическую активность определяют (см. 2.3.3.) на основании кинетических экспериментов и представлений о механизме каталитических реакций (см. 2.3.2).
Как правило, каталитическая активность смеси катализаторов не аддитивна. Часто она значительно превосходит активность отдельно взятых катализаторов.
Увеличение каталитической активности наблюдается при добавлении к катализатору некоторых веществ, называемых промоторами, влияющих на структуру поверхности катализатора и тем самым способствующих ускорению каталитической реакции. Такой катализатор называют смешанным, или промотированным. Примером сказанному может служить каталитическая реакция окисления SO2 , скорость которой увеличивается в сотни раз, когда к катализа-
тору V2O5 добавляют небольшое количество промотора – сульфатов щелочных
металлов.
Вещества, снижающие при добавлении к катализатору его активность или полностью исключающие каталитическое действие, называют каталитиче-
173

Х и м и ч е с к а я к и н е т и к а
скими ядами. К ним относятся, например, соединения Sb, P, Pb, As, Hg . Ме-
таллические катализаторы отравляются соединениями кислорода (например H2O, CO ) и серы (например CS2 , H2 S ). Эти яды образуют с катализаторами
более прочную химическую связь, чем реагирующие вещества. В ферментативном катализе для обозначения этого явления применяют термины – ингибирование и ингибитор, т.е. говорят об ингибировании катализатора.
Специфичность катализатора заключается в том, что реакции данного типа ускоряются катализаторами лишь определенного химического состава независимо от того, являются они гомогенными или гетерогенными. Например, кислотно-основные реакции ускоряются кислотами и основаниями, а окисли- тельно-восстановительные – переходными металлами или их соединениями. Следует отметить, что даже в пределах одной группы периодической системы элементов каталитические свойства меняются немонотонно. Например, из всех переходных элементов восьмой группы лишь железо обладает достаточно высокой активностью в реакции синтеза аммиака. Специфичность свойственна в той или иной мере всем катализаторам и обусловлена специфичностью химических связей, возникающих между катализатором и исходным веществом (реагентом). Лишь определенные группы атомов катализатора, называемые каталитическим или активным центром, участвуют в образовании химической связи с реагентом. В гомогенном катализе каждая молекула катализатора может рассматриваться как активный центр. В комплексных соединениях переходных металлов в качестве активных центров может выступать «вакантное место» в координационной сфере комплексного соединения иона металла. При гетерогенном катализе активные центры находятся на поверхности твердого тела (катализатора) и представляют собой один или группу атомов, ионов кристаллической решетки. Более сложное строение имеют активные центры ферментов. Специфичность каталитического действия того или иного катализатора определяется химическим составом, строением и структурой его активных центров. Специфичность катализатора проявляется по-разному: от способности изменять скорость превращения нескольких классов соединений до отдельных химических веществ. Так, например, кислоты являются катализаторами для многих классов химических соединений, в то время как ферменты ускоряют лишь определенный биохимический процесс, т.е. высокоспецифичны.
Селективность катализатора – это свойство катализатора ускорять химическое превращение лишь в одном из нескольких возможных направлений. Так, например, в присутствии оксида алюминия при 700 K происходит увели-
чение скорости дегидратации этилового спирта:
C2 H5OH →C2 H4 + H2O ,
а в присутствии металлической меди – дегидрирования:
C2 H5OH →C2 H4O + H2 .
174

Х и м и ч е с к а я к и н е т и к а
При этом следует заметить, что в отсутствие катализатора эти реакции являются параллельными.
Селективность катализатора может быть охарактеризована долей исходного вещества, превратившегося в целевой продукт (интегральная селективность), или отношением скорости образования целевого продукта к сумме скоростей химического превращения исходных веществ по всем возможным на-
правлениям (дифференциальная селективность). Так, если в реакции
A + B →Q + R
целевым продуктом является вещество Q, то дифференциальная селективность катализатора равна
CK =vQ / (vA +vB ) |
(2.386) |
где vQ , vA и vB – скорости каталитической реакции по отношению к веществам
Q,, A и В.
Селективность обусловлена природой (составом) катализатора, а также зависит от пористости, размеров зерен и их упаковки. Селективность катализатора зависит от условий проведения и степени завершенности реакции. Селективность является важным технологическим свойством катализатора. Ее повышение позволяет уменьшить количество побочных веществ, а значит, путем подбора подходящего катализатора можно увеличить выход целевого (нужного) продукта. Наибольшей селективностью (95–100%) обладают ферменты и некоторые катализаторы, используемые в гомогенном катализе. Катализаторы, применяемые в гетерогенном катализе, как правило, обладают более низкой селективностью ( 70%).
2.3.2. Механизмы каталитических реакций
Увеличение скорости каталитических реакций может происходить за счет снижения энергии активации и увеличения энтропии активации, что явствует из теории абсолютных скоростей реакций [см., в частности, уравнения (2.374),
(2.382)].
Для оценки влияния катализатора на энергию реакции используют понятие степень компенсации. При химических реакциях образование новых химических связей требует разрыва определенных связей в исходных соединениях, что требует затраты энергии. Энергия активации обычно значительно меньше энергии, затрачиваемой на разрыв химических связей. Это обусловлено тем, что при движении по пути реакции (через переходное состояние) часть энергии, требуемой для разрыва старых связей, компенсируется энергией, освобожденной при образовании новых. Степень компенсации κ определяет реакционную способность веществ в рассматриваемой реакции
175

Х и м и ч е с к а я к и н е т и к а
κ = |
∑Di − Ea |
, |
(2.387) |
∑Di |
|||
где ∑Di – сумма энергий разрываемых связей. При Ea =0 |
получаем κ =1, и |
||
степень компенсации полная. При |
Ea = ∑Di |
κ = 0 , т.е. компенсация отсутст- |
|
вует. Для реакций между стабильными молекулами (без участия свободных радикалов или атомов) степень компенсации обычно не превышает 70%. Взаимодействуя с реагирующими веществами, входя в состав активированного комплекса, катализатор увеличивает степень компенсации, снижает энергию активации и тем самым увеличивает скорость химического превращения.
Рассмотрим указанные причины увеличения скорости каталитических реакций применительно к двум механизмам каталитических реакций – стадий-
ному и слитному.
Стадийный (раздельный, диссоциативный) механизм каталитических ре-
акций заключается в замене одной каталитической реакции на несколько стадий последовательного взаимодействия исходных веществ с катализатором с возможным образованием на каждой стадии активированного комплекса.
При катализе бимолекулярная реакция типа
A + B →Q + R |
(2.388) |
может протекать в следующие две стадии: |
|
A + K →(AK )≠ → AK, AK + B →(ABK )≠ →Q + R + K , |
(2.389) |
где AK – устойчивое промежуточное соединение с катализатором; |
(AK )≠ , |
(ABK )≠ – промежуточные активированные комплексы. |
|
Согласно слитному (ассоциативному, синхронному) механизму в процес-
се реакции происходит одновременное взаимодействие с катализатором всех исходных веществ и образование одного активированного комплекса:
A + B + K →(ABK )≠ →Q + R + K . |
(2.390) |
Таким образом, если в стадийных каталитических реакциях, например (2.389), образуется, по крайней мере, не меньше двух активированных ком-
плексов, то в слитных каталитических реакциях, например (2.390), невозможно
возникновение более одного активированного комплекса.
Профили ППЭ вдоль координаты реакции в каталитических реакциях, идущих по стадийному и слитному механизму, представлены на рис. 2.42.
176
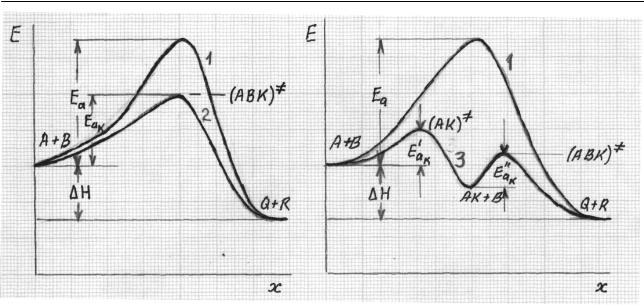
Х и м и ч е с к а я к и н е т и к а
Рис. 2.42. Профили ППЭ вдоль координаты реакции x:
1 – для некаталитического процесса, идущего в соответствии с уравнением (2.388); 2 – для каталитической реакции со слитным механизмом согласно
уравнению (2.390) ( ∆H – тепловой эффект реакции); 3 – для каталитической реакции со стадийным механизмом по уравнению (2.389)
В случае слитного механизма [рис. 2.42 (а)] как некаталитическая, так и каталитическая реакции (кривые 1 и 2) идут с преодолением одного потенциального барьера, но с существенным снижением энергии активации для каталити-
ческой реакции (Eaк < Ea ). Таким образом, фактором, определяющим увеличе-
ние скорости реакции при слитном механизме, является уменьшение энергии активации и соответственно повышение степени компенсации при химическом взаимодействии реагентов с катализатором. Энтропия активации в каталитической реакции по слитному механизму уменьшается, способствуя понижению константы скорости реакции. Однако превалирующим фактором остается понижение энергии активации, определяющее, как уже отмечалось, увеличение скорости реакции.
В случае стадийного механизма каждая стадия реакции [рис. 2.42 (б)] имеет свою энергию активации Ea′к и Ea′′к . Для реакции, протекающей по механизму
(2.389), профиль ППЭ (кривая 3) характеризуется наличием двух максимумов, соответствующих активированным комплексам отдельных стадий (AK )≠ ,
(ABK )≠ и минимума, соответствующего продукту промежуточного взаимодействия AK+B. Энергия активации всей каталитической реакции равна
∑Ea |
= Ea′ |
+ Еа′′ . |
(2.391) |
к |
к |
к |
|
177

Х и м и ч е с к а я к и н е т и к а
Если суммарная энергия всех стадий каталитической реакции ∑Еа |
к |
меньше |
|
|
энергии активации некаталитической реакции Еа , т.е. ∑Еак < Еа , то увеличе-
ние скорости химической реакции происходит за счет роста доли активных молекул. Однако суммарная энергия активации каталитической реакции не всегда снижается по сравнению с энергией активации реакции без катализатора – воз-
можны случаи, когда ∑Еак > Еа . В этих условиях фактором, определяющим
увеличение скорости реакции, является повышение энтропии активации Для серии однотипных катализаторов в одной и той же реакции возможен
переход от диссоциативного к синхронному механизму. При этом наблюдается уменьшение энергии и энтропии активации при переходе от одного катализатора к другому. Первый фактор будет приводить к увеличению, а второй – к уменьшению константы скорости реакции. Следовательно, константа скорости реакции будет меняться меньше, чем в том случае, когда практически изменяется лишь один из кинетических параметров реакции (энергия или энтропия активации). В этом случае будет наблюдаться компенсационный эффект. Если между энергией и энтропией активации имеет место линейное соотношение
∆S0≠ =α + βE , |
(2.392) |
aк
где α, β – постоянные коэффициенты в ряду сходных катализаторов для дан-
ной реакции, то при некоторой температуре скорость реакции на всех катализаторах данной серии будет одинакова. Такая температура называется изокинетической. Компенсационный эффект наблюдается как при гомогенном, так и гетерогенном катализах. Однако в каждом отдельном случае причины его появления могут быть различны.
2.3.3. Скорость и причины каталитических реакций
Скорость гомогенных и гетерогенных каталитических реакций определяется через каталитическую активность Ак . Если скорость каталитической ре-
акции обозначить через vк , а скорость той же реакции в отсутствие катализатора через v0 , то для гетерогенного катализа активность равна
Aк =vк −v0 (1 −ϕ), |
(2.393) |
где ϕ – доля объема системы, занимаемая катализатором и недоступная для
реагирующих веществ.
Величина каталитической активности показывает, насколько увеличивается скорость гетерогенной каталитической реакции по сравнению со скоростью этой же реакции в отсутствии катализатора.
Для гетерогенного катализа широко используют удельную каталитическую активность, т.е. каталитическую активность, отнесенную к массе ката-
178

Х и м и ч е с к а я к и н е т и к а
лизатора am , к единице объема катализатора aV и к единице площади поверхности катализатора aS , определяемую по следующим выражениям:
|
|
A |
|
|
vк −v0 |
(1 −ϕ) |
|
(2.394) |
||
a |
= |
к |
= |
|
|
|
, |
|||
|
|
|
|
|
|
|||||
m |
|
mк |
|
|
mк |
|
|
|||
|
|
|
|
|
|
|||||
|
|
A |
|
vк −v0 |
(1 −ϕ) |
|
(2.395) |
|||
a |
= |
к |
|
= |
|
|
|
, |
||
|
|
|
|
|
||||||
V |
|
Vк |
|
|
Vк |
|
|
|||
|
|
|
|
|
|
|||||
|
|
A |
|
vк −v0 |
(1 −ϕ) |
|
(2.396) |
|||
aS |
= |
к |
|
= |
|
|
|
, |
||
Sк |
|
Sк |
||||||||
|
|
|
|
|
|
|||||
где mк, Vк, Sк – масса, объем и площадь поверхности катализатора соответственно.
При гомогенном катализе ϕ →1 и величиной v0 (1 −ϕ) можно пренебречь. Тогда формула (2.393) упрощается:
Aк =vк . |
(2.397) |
Для гомогенного катализа удельную каталитическую активность ac определя-
ют в расчете на число молей катализатора в единице объема жидкой или газовой среды cк . Вместо удельной каталитической активности ac при этом часто
используют тождественное, с учетом выражения (2.397), понятие – число оборотов катализатора nк . Дело в том, что, как уже было показано (см. 2.3.2),
любая каталитическая реакция включает, по крайней мере, две стадии: 1. взаимодействие активного центра с молекулой реагента и образование активированного комплекса, 2. распад активированного комплекса с образованием продуктов реакции и «свободного» активного центра. Последний вновь вступает во взаимодействие с молекулами исходных веществ. Такие циклы могут повторяться многократно. Число циклов, совершающихся за единицу времени на одном активном центре, называется числом оборотов катализатора. Число оборотов катализатора служит мерой каталитической активности. Его легко подсчитать, если разделить скорость реакции на молярную концентрацию катализатора. С учетом всего сказанного выше для удельной каталитической активности при гомогенном катализе справедливо следующее выражение:
|
ac ≡ nк = Aк / ск =vк / ск , |
(2.398) |
||
где n |
– число оборотов катализатора, c−1 ; v |
к |
– скорость каталитической реак- |
|
к |
|
|
|
|
ции, моль/ (м3 с); ск – молярная концентрация катализатора, |
моль/ м3 . |
|||
179

Х и м и ч е с к а я к и н е т и к а
Для катализаторов кислотно-основного действия при 298 К число оборотов составляет 10−7 −10−2 с−1 , для комплексов переходных металлов – 1 −104 с−1 , а для ферментов – 102 −105 с−1.
Активность катализатора, также как и скорость реакции, зависит от условий проведения каталитического процесса: температуры, концентрации реагентов, давления, а также растворителя, если каталитическая реакция протекает в жидкой фазе. Поэтому сравнение каталитической активности различных катализаторов проводят при строго одинаковых условиях.
При стадийном механизме гомогенная каталитическая бимолекулярная реакция (2.388) протекает в две стадии (2.389), причем продукт первой стадии
AK является исходным веществом для второй. Таким образом, мы имеем последовательную реакцию. Известно (см.2.2.1.5.А), что для последовательных реакций кинетические уравнения зависят только от константы скорости лимитирующей стадии. Поэтому в случае, когда лимитирующей является первая стадия, скорость каталитической реакции равна
v |
к |
=v = − |
dcA |
= k c |
c |
K |
, |
(2.399) |
|
||||||||
|
1 |
dτ |
1 A |
|
|
|
||
|
|
|
|
|
|
|
|
|
где v1 – скорость первой стадии реакции, |
k1 – константа скорости первой ста- |
|||||||
дии реакции; cA , cK – концентрации исходного вещества и катализатора. Сразу же отметим, что уравнением аналогичного типа будет определяться и скорость гомогенной каталитической реакции (2.388) при ее протекании по слитному
механизму (2.390)
vк = − |
dcA |
= kcAcBcK . |
(2.400) |
|
|||
|
dτ |
|
|
В случае, когда лимитирующей является вторая стадия, скорость каталитической реакции равна
vк =v2 = − |
dcAK |
= k2cAK cB |
(2.401) |
|
dτ |
||||
|
|
|
||
или, с учетом K1 = cAK / cAcK , имеем |
|
|
||
vк = k2 K1cAcK cB , |
(2.402) |
|||
где v2 – скорость второй стадии реакции; |
k2 – константа скорости второй ста- |
|||
дии реакции; cA , cB , cK и cAK – концентрации исходных веществ, катализатора и устойчивого промежуточного соединения с катализатором; K1 – константа равновесия первой стадии реакции.
180