

Физ химия и физ т.д
..pdfХ и м и ч е с к а я к и н е т и к а
|
|
4πr2 |
с − с |
S |
|
(2.426) |
|||
vвн = D |
|
|
г |
|
|
|
, |
||
|
4 |
πr |
3 |
δ |
|
||||
|
|
3 |
|
|
|
|
|
|
|
|
|
г |
|
|
|
|
|||
из которого после элементарных преобразований получаем выражение для скорости диффузии реагента к внешней поверхности гранулы (внешней диффузии):
v |
вн |
= 3D (c − c |
S |
). |
(2.427) |
|
δr |
|
|
||
|
|
г |
|
|
|
Обозначив 3D /δrг через kвн, выражение (2.427) |
можно переписать в виде |
||||
vвн = kвн (c − cS ), |
(2.428) |
||||
где kвн – константа внешнего массообмена (константа массообмена между
внешней поверхностью катализатора и объемом газовой или жидкой фазы). Если внешняя диффузия является лимитирующей стадией гетерогеннокаталитического процесса, то сS =0 , а градиент концентрации достигает мак-
симального значения. В этом случае, исходя из (2.428), имеем
vвн = kвнc , |
(2.429) |
т.е. скорость внешней диффузии, а, следовательно, и скорость всего каталитического процесса пропорциональна концентрации реагента. При уменьшении размера гранул катализатора и толщины диффузионного слоя наблюдается увеличение константы внешнего массообмена. Увеличение скорости движения жидкости или газа относительно твердых частиц катализатора приводит к уменьшению толщины диффузионного слоя. Исходя из этого, можно заключить, что скорость внешней диффузии будет тем больше, чем выше скорость протекания газа или жидкости через слой катализатора (в проточных реакторах) или интенсивность перемешивания суспензии катализатора в жидкой фазе (в непроточных реакторах).
Диффузия реагирующих веществ в пористом катализаторе протекает за счет разности концентраций на внешней поверхности сS и в объеме пористой гра-
нулы cV (см. рис. 2.44). Примем, что диффузия в пористом катализаторе подчиняется уравнению Фика (2.411) с тем различием, что вместо истинного вво-
дится эффективный коэффициент диффузии D . Данный коэффициент, ис-
ходя из того, что диффузия протекает не по всему объему гранулы катализатора, а лишь в объеме пор, зависит от пористости катализатора ε , равной объему пор в единице объема пористой системы, и от степени извилистости пор в грануле катализатора, называемой фактором извилистости ι :
191

Х и м и ч е с к а я к и н е т и к а
D = D |
ε |
= DП, |
(2.430) |
|
ι |
|
|
где П =ε /ι – проницаемость пористой гранулы катализатора, которая для большинства катализаторов лежит в пределах 0,1–0,3.
Одновременно с процессом диффузии реагентов в гранулу катализатора на стенках пор протекает каталитическая реакция. Пусть ее скорость на равнодоступной поверхности выражается уравнением первого порядка
vк = kcV . |
(2.431) |
Отнеся эту скорость к единице объема пористого катализатора Vк
vк |
= |
k |
с |
(2.432) |
|
||||
V |
V |
V |
|
|
к |
|
к |
|
|
и обозначив vк /Vк через vS , а k /Vк через kS , получим |
|
|||
vS |
= kS cV . |
(2.433) |
||
На рис. 2.44 наглядно видно уменьшение концентрации реагента от внешней поверхности к центру гранулы катализатора, вызывающее соответствующее снижение скорости реакции в центральной части катализатора. Этот эффект суммарно будет выражаться в том, что отнесенная к единице объема гранулы катализатора скорость реакции, наблюдаемая на опыте vн , будет значи-
тельно ниже vS . Лишь в случае, когда по всему объему гранулы катализатора cV =cS ,
vн =vS = kS cS . |
|
Понятно, что при cV <cS всегда vн <vS . Величина |
|
η = vн |
(2.434) |
vS |
|
характеризует степень использования внутренних слоев гранулы катализатора и называется фактором эффективности или степенью использования поверхности.
Уравнение макрокинетики (в случае гранул катализатора сферической формы) имеет вид
v |
н |
= |
3 |
k |
S |
D c |
S |
. |
(2.435) |
r |
|||||||||
|
|
|
г |
|
|
|
|
|
|
Из уравнения (2.435) следует, что скорость процесса уменьшается с увеличе-
нием размера гранул катализатора и зависит как от kS , так и от D . Порядок реакции при этом остается равным единице.
192
Х и м и ч е с к а я к и н е т и к а
Наличие сильного диффузионного торможения оказывает влияние не только на скорость реакции, но и на величину энергии активации каталитического процесса. Наблюдаемая на опыте энергия активации каталитического процесса в условиях сильного диффузионного торможения равна половине энергии активации той же реакции, протекающей на равнодоступной поверхности.
Следует отметить, что если реакция на равнодоступной поверхности подчиняется уравнению n-го порядка vS = kS cVn , то согласно теоретическим поло-
жениям макрокинетики скорость процесса при наличии сильного диффузионного торможения может быть выражена уравнением
|
|
= |
3 |
k |
|
D c |
n+1 |
(2.436) |
||
v |
н |
S |
2 |
. |
||||||
r |
||||||||||
|
|
|
S |
|
||||||
|
|
|
г |
|
|
|
|
|
|
|
Из (2.436) следует, что при увеличении диффузионного торможения порядок реакции изменяется от единицы до (n +1)/ 2 . Так, для реакции второго порядка
при наличии сильного диффузионного торможения порядок, наблюдаемый на опыте, будет равен 1,5.
С учетом всего вышесказанного сформулируем признаки протекания гете- рогенно-каталитической реакции в той или иной области.
Признаками протекания реакции во внешнедиффузионной области являются: 1. зависимость скорости процесса от интенсивности перемешивания жидкой фазы или от скорости протекания газа (жидкости) через слой катализатора (поскольку kвн зависит от δ ); 2. увеличение скорости процесса с уменьшением
размера гранул катализатора; 3. близость энергии активации реакции к энергии активации диффузии (4–8 кДж/моль).
Признаками протекания реакции во внутридиффузионной области являются: 1.увеличение скорости реакции с уменьшением размера гранул катализатора при одновременном отсутствии ее зависимости от скорости потока реагента или интенсивности перемешивания жидкой фазы; 2. изменение порядка реакции при увеличении размера гранул катализатора, если n >1; 3. зависимость скорости реакции от пористости, радиуса гранул и степени извилистости пор катализатора.
При протекания реакции в кинетической области (равнодоступная поверхность) ее скорость не зависит от интенсивности перемешивания реакционной смеси, скорости потока газа или жидкости через слой катализатора, размера гранул катализатора и его пористости.
При изменении условий проведения каталитического процесса может происходить переход реакции из одной области в другую. Так, по мере понижения температуры протекание реакции переходит из внешнедиффузионной области через внутридиффузионную в кинетическую область. К переходу из диффузионной области в кинетическую приводит и измельчение гранул катализатора.
193

Х и м и ч е с к а я к и н е т и к а
Процесс перехода реакции из диффузионной области в кинетическую приводит к увеличению скорости реакции и энергии ее активации.
2.3.4. Ферментативный катализ
Катализ под действием ферментов называют ферментативным. Ферменты, или энзимы – это биокатализаторы, продукты жизнедеятельности живых организмов. Они имеют более сложное строение по сравнению с неорганическими катализаторами. Ферменты бывают простые и сложные. Простые, или однокомпонентные, ферменты состоят только из белковых тел, в то время как сложные ферменты включают белковую и небелковую составляющую. При этом небелковая составляющая резко увеличивает каталитическую активность фермента.
По сравнению с химическим катализом ферментативный имеет ряд особенностей, к числу которых относятся высокая каталитическая активность, четкая специфичность и селективность, повышенное влияние окружающей среды.
Каталитическая активность биологических катализаторов в миллионы раз превосходит активность химических. При протекании одних и тех же реакций с использованием обычного катализатора и фермента наблюдается колоссальное увеличение скорости ферментативного катализа (на 12–13 порядков!). Так, например, константа скорости реакции гидролиза мочевины под действием обыч-
ного кислотного катализатора (H3O+ ) |
равна 7,4 10−7 л/ (моль с), в то время |
||
как под действием фермента уреазы – |
5,0 106 л/ (моль с). Данный факт обу- |
||
словлен также снижением энергии |
активации |
за счет |
фермента (от |
78,4 кДж/ мольдо 27,2 кДж/ моль в приведенном |
примере). |
Скорость реак- |
|
ций, катализируемых ферментами, зависит от концентрации реагирующих веществ и условий среды. Ускорение ферментативной реакции1 происходит, в частности, благодаря тому, что в системе создаются условия для автокаталитической реакции.
Ферменты, в отличие от химических катализаторов обладают большей специфичностью: каждый из них способен ускорить только строго определенную реакцию и даже образование определенных стереоизомеров. Реже ферменты катализируют группы реакций: например, фермент пепсин способствует расщеплению белков. Ферменты более чувствительны к изменению внешних условий, в частности температуры, чем неорганические катализаторы. Для большинства растительных ферментов температурный интервал составляет
40 −60 oC , а для животных ферментов – 40 −50 oC . Уже при температуре 70 −80 oC происходит необратимое разрушение ферментов вследствие денатурации белка.
1 Вопросы теории ферментативного катализа весьма подробно рассмотрены в [12]. 194

Х и м и ч е с к а я к и н е т и к а
Свою активность ферменты проявляют при строго определенном значении pH среды. Например, для уже упоминавшегося пепсина оптимальное значение pH составляет 1,5–2,0.
Ферментативный каталитический процесс обусловлен способностью к взаимным синхронным изменениям структур фермента и субстрата (субстрат – вещество на которое воздействует фермент). Эти изменения связа-
ны с конформаццией1 взаимодействующих макромолекул.
Специфичность взаимодействия фермента и субстрата определяется приципом ключ–замок [1]. Подобно ключу, отпирающему определенный замок, фермент оказывает каталитическое действие на определенный субстрат. Одна из причин такой специфичности фермента заключается в том, что в белковой молекуле фермента каталитические функции выполняют отдельные небольшие участки – активные центры, представляющие собой ориентированные в пространстве функциональные группы в основном белковых молекул. Структура активного центра может возникнуть в процессе сближения молекул фермента и субстрата.
Активные центры способны к избирательной адсорбции молекул субстрата, в результате чего образуется единый комплекс, внутри которого происходит химическая реакция между ферментом и субстратом. Возникновение в комплексе своеобразной микрофазы с параметрами (диэлектрическая проницаемость, полярность и др.), отличными от параметров среды, способствует ускорению химической реакции. Процесс завершается десорбцией продуктов реакции и регенерацией активных центров для новых каталитических актов. Большое число активных центров способствует протеканию каталитического процесса в большой массе субстрата.
Действие всех факторов обусловлено и геометрическим соответствием структур активного центра фермента и самого субстрата (ключ–замок).
Простейшая схема ферментативной реакции может быть изображена следующим образом:
→1 |
k2 |
|
k |
|
(2.437) |
E + S ← ES →E + P , |
||
k−1 |
|
|
где k1, k−1, k2 – константы скорости реакции. В соответствии с (2.437) |
фермент |
|
E обратимо реагирует с субстратом S с образованием комплекса ES , который |
||
распадается на продукты реакции Р и исходный фермент Е. |
|
|
Применение квазиравновесного |
приближения (см. 2.2.1.5.В) |
к схеме |
(2.437), что возможно при условии k2 |
k−1 , с учетом уравнения материально- |
|
го баланса
cE =c0E −cES ,
1 Коформациями называют энергетически неравноценные формы макромолекул (в данном случае фермента и субстрата), возникающие при повороте мономерных звеньев без разрыва химических связей.
195

Х и м и ч е с к а я к и н е т и к а
где cOE , cE –начальная и текущая концентрации фермента соответственно; cES
– текущая концентрация комплекса, позволяет выразить скорость образования продукта (его текущая концентрация cP ) через начальную концентрацию фер-
мента и текущую концентрацию субстрата cS :
v = |
dcP |
|
= |
|
k2c0E cS |
. |
(2.438) |
|||
dτ |
|
|
|
|
||||||
|
|
|
|
KS + cS |
|
|||||
Входящая в (2.438) константа KS , называемая субстратной константой, оп- |
||||||||||
ределяется выражением |
|
|
|
|
|
|
|
|
||
KS = |
k−1 |
|
= |
cE cS |
. |
(2.439) |
||||
|
|
|||||||||
|
|
|
k1 |
|
cES |
|
||||
При увеличении концентрации субстрата скорость реакции, исходя из выраже- |
|
ний (2.438), (2.439), стремится к предельному значению: |
|
vmax = k2c0E . |
(2.440) |
Подставляя (2.440) в(2.438), получаем следующее соотношение, связывающее скорость реакции с максимальной скоростью реакции:
v = |
dcP |
= |
vmax cS |
. |
(2.441) |
dτ |
|
||||
|
|
KS + cS |
|
||
Определение параметров KS и vmax обычно проводят на основании измерения
зависимости начальной скорости от начальной концентрации субстрата для ряда начальных концентраций.
Наиболее часто для анализа кинетических схем ферментативного катализа
используют метод стационарных концентраций (см. 2.2.1.5.В), что допустимо при условии k2 k1 . Применение этого метода к простейшей схеме катализа
(2.437) дает уравнение Михаэлиса-Ментен:
v = |
dcP |
= |
vmax cS |
, |
(2.442) |
dτ |
|
||||
|
|
KM + cS |
|
||
где vmax – максимальная скорость реакции (при бесконечно большой концентрации субстрата), отвечающая выражению (2.440); KM – константа Михаэлиса, определяемая выражением
KM |
= |
k2 + k−1 |
. |
(2.443) |
|
||||
|
|
k1 |
|
|
Константа Михаэлиса соответствует концентрации субстрата, при которой скорость реакции равна половине максимальной скорости. Типичные значения KM
196

Х и м и ч е с к а я к и н е т и к а
лежат в пределах от 10−6 до 10-1 моль/ л.Константу скорости k2 иногда называют числом оборотов фермента. Она может изменяться в пределах от1,7 10−1 до 1,7 106 c−1 .
Уравнение (2.442) можно записать в координатах, более удобных для об-
работки экспериментальных данных: |
|
|
|
|
|
|
|
|
|
|
|
||||
1 = |
|
1 |
|
+ |
KM |
|
|
1 |
|
(2.444) |
|||||
v |
|
|
|
|
|
|
c |
||||||||
v |
max |
|
v |
max |
|
|
|
||||||||
|
|
|
|
|
|
|
S |
|
|||||||
или |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
v =v |
max |
− K |
|
|
v |
. |
(2.445) |
||||||||
|
|
|
|||||||||||||
|
|
|
|
|
|
M c |
S |
|
|||||||
|
|
|
|
|
|
|
|
|
|
|
|
||||
Для определения параметров KM и vmax |
по уравнениям (2.444) |
и (2.445) про- |
|||||||||||||
водят серию измерений начальной скорости реакции v0 от начальной концентрации субстрата c0S и представляют экспериментальные данные в координатах 1/v0 = f (1/ c0S ) или v0 = f (v0 / c0S ).
2.3.5. Примеры и задачи
А. Примеры решения задач Пример 1. Найдите константу Михаэлиса и максимальную скорость гидро-
лиза аденозинтрифосфата, катализируемого минозином по кинетическим данным, приведенным на рис. 2.45.
Рис. 2.45. Кинетические данные
197

Х и м и ч е с к а я к и н е т и к а
Решение
Уравнение Михаэлиса-Ментен в координатах, приведенных на рис. 2.45, имеет вид (2.445). Следовательно, точки пересечения с осями имеют следую-
щие координаты: (0; vmax ) для оси ординат и (vmax / KM ; 0) для оси абсцисс. Точка пересечения с осью ординат дает значения vmax = 2,1 10−6 моль/ (л с).
Точка пересечения с осью абсцисс позволяет найти константу Михаэлиса: vmax / KM =14,6 10−3 c−1 , откуда KM = 2,1 10−6 /14,6 10−3 =1,44 10−4 моль/ л.
Пример 2. Для некоторой ферментативной реакции константа Михаэлиса
равна 0,035 моль/ л. Скорость реакции |
при |
концентрации |
субстрата |
0,110 моль/ л равна 1,15 10−3 моль/ (л с). |
Найдите |
максимальную |
скорость |
этой реакции. |
|
|
|
Решение
Исходя из уравнения (2.445), выражение для максимальной скорости имеет следующий вид:
vmax =v + KM cvS .
Подставляя в это выражение конкретные значения величин, получаем
vmax =1,15 10−3 + 0,0351,15 10−3 =1,52 10−3 моль/ (л с). 0,110
В. Задачи для самостоятельного решения Задача 1. Начальная скорость окисления сукцината натрия в фумарат на-
трия под действием фермента сукциноксидазы была измерена для ряда концентраций субстрата:
сS , моль/ л |
0,01 |
0,002 |
0,001 |
0,0005 |
0,00033 |
|
|
|
|
0,62 |
|
|
|
|
|
|
|
v 106 , моль/ (л с) |
1,17 |
0,99 |
0,79 |
0,50 |
|
|
|
|
|
|
|
|
|
|
|
|
|
Определите константу Михаэлиса данной реакции.
Ответ: 4,86 10−4 моль/ л.
198
Х и м и ч е с к а я к и н е т и к а
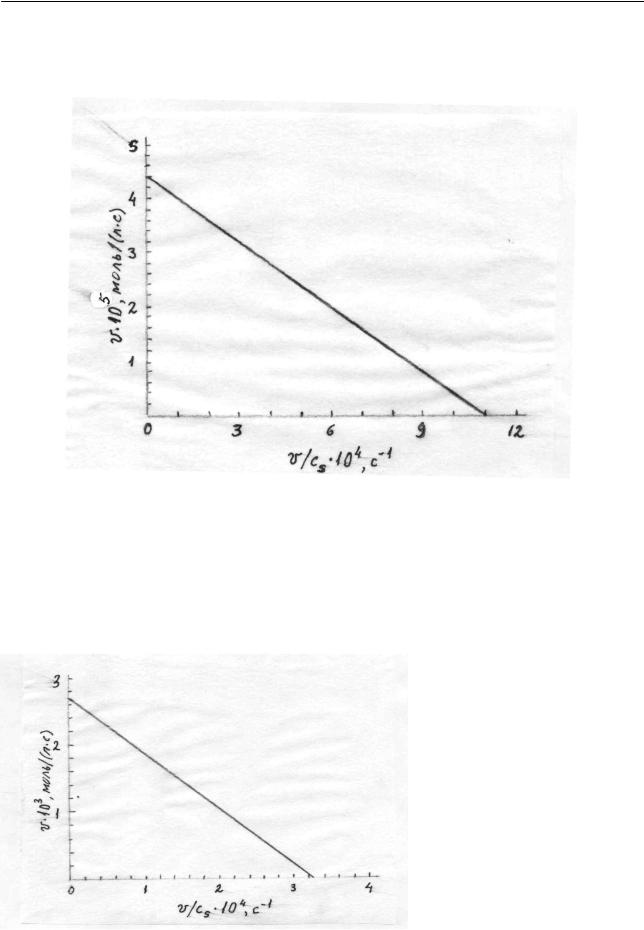
Задача 2. Найдите константу Михаэлиса и максимальную скорость каталитического разложения гидроперекиси по кинетическим данным, приведенным на рис. 2.46.
Рис. 2.46. Кинетические данные
Ответ: KM =0,040 моль/ л; vmax = 4,4 10−5 моль/ (л с).
Задача 3. Найдите константу Михаэлиса и максимальную скорость каталитического окисления циклогексена трет-бутилпероксидом по кинетическим данным, приведенным на рис. 2.47.
Рис. 2.47. Кинетические данные
199

Х и м и ч е с к а я к и н е т и к а
Ответ: KM =0,083 моль/ л; vmax = 2,7 10−5 моль/ (л с).
ЛИТЕРАТУРА
1.Зимон А.Д., Лещенко Н.Ф. Физическая химия: Учеб. для вузов. – М. Хи-
мия, 2000. – 320 с.
2.Физическая химия. Ч. 1. Химическая термодинамика: Текст лекций / Зенин Г.С., Сысоева В.В., Привалова Т.А., Пенкина Н.В. – СПб.: СЗПИ, 1997. – 60 с.
3.Киреев В.А. Курс физической химии: Учеб. для вузов. 3-е изд, перераб. и
доп. – М.: Химия, 1975. – 776 с.
4.Стромберг А.Г., Семченко Д.П. Физическая химия: Учеб. для хим. спец. вузов / Под ред. А.Г.Стромберга. – 3-е изд., испр. и доп. – М.: Высш. шк., 1999. – 527 с.
5.Физическая химия. В 2 кн. Кн. 2. Электрохимия. Химическая кинетика и катализ: Учеб. для вузов / К.С. Краснов, Н.К.Воробьев, И.Н. Годеев и др.; Под ред. К.С. Краснова – 2-е изд., перераб. и доп. – М.: Высш. шк., 1995. – 319 с.
6.Эткинс П. В 2 т. Т. 2. Физическая химия / Пер. с англ. / Под ред. К.П Бу-
тина. – М.: Мир, 1980. – 584 с.
7.Даниэльс Ф., Ольберти Р. Физическая химия / Пер. с англ. / Под ред.
К.В. Топчиевой. – М.: Мир, 1978. – 645 с.
8.Кноре Д.Г., Крылова Л.Ф., Музыкантов В.С. Физическая химия: Учеб. для биол. ф-тов университетов и пед. вузов. 2-е изд, испр. и доп. – М.: Высш.
шк., 1990. – 416 с.
9.Еремин Е.Н. Основы химической кинетики: Учеб. пособие для университетов и химико-технологических вузов. 2-е изд., доп.. – М.: Высш. шк., 1976. – 375 с.
10.Жуховицкий А.А., Шварцман Л.А. Физическая химия: Учеб. для вузов. 4-е изд., перераб. и доп. – М.: Металлургия, 1987. – 688 с.
11.Алексеев А.И. Кинетические расчеты технологических процессов в производстве минеральных удобрений и глинозема: Учеб. пособие. – Л.: СЗПИ, 1986. – 80 с.
12.Денисов Е.Т., Саркисов О.М., Лихтенштейн Г.И. Химическая кинетика: Учеб. для вузов. – М.: Химия, 2000. – 568 с.
13.Сена Л.А. Единицы физических величин и их размерности: Учебносправочное руководство. 3-е изд., перераб. и доп. – М.: Гл. ред. физ.-мат. лит., 1988. – 432 с.
14.Чертов А.Г. Единицы физических величин: Учеб. пособие для вузов. –
М.: Высш. шк., 1977. – 287 с.
15.Бурлешин А.В., Зенин Г.С. Сраго И.А. Физическая химия: Метод.указания к выполнению лабораторной работы «Исследование кинетики взаимодействия малахитового зеленого с гидроксид-ионом. – СПб.: СЗПИ, 1996. – 16 с.
200