

Физ химия и физ т.д
..pdfХ и м и ч е с к а я к и н е т и к а
и из (2.146)
c2 = K2c3 = |
c01K2 |
(1 |
− e |
−k τ |
). |
(2.150) |
|
1 |
|||||
K2 +1 |
Таким образом, для случая k1 k2 уравнения кинетических кривых зависят
только от константы скорости первой стадии, являющейся лимитирующей. Поскольку промежуточное вещество P находится в равновесии с веществом B, к концу реакции оно остается в системе.
В рассматриваемом случае, начиная с некоторого момента времени (после достижения максимума на кинетической кривой промежуточного продукта) в
уравнении (2.130), членом e−k2τ по сравнению с членом e−k1τ |
можно пренеб- |
||
речь. Тогда, с учетом k2 − k1 k2 , имеем |
|
||
c3 |
c01k1 |
e−k1τ . |
(2.151) |
|
|||
|
k2 |
|
|
Скорость превращения промежуточного вещества P в B будет значительно выше скорости его возникновения из A, а его концентрация будет весьма низкой. Отношение c3 / c1 можно найти из (2.129) и (2.151):
c |
|
(k1 / k2 )c01e−k1τ |
|
k |
|
(2.152) |
||
3 |
|
|
|
|
1 |
const. |
||
c |
c e−k1τ |
k |
2 |
|||||
|
|
|
|
|||||
1 |
|
01 |
|
|
|
|
||
Таким образом, отношение c3 / c1 с течением времени не изменяется. Аналогич-
ная ситуация (рис. 2.12) возникает при проведении реакции в стационарных условиях (см. 2.2.1.5.В).
Рис. 2.12. Зависимость концентрации промежуточного вещества от времени для последовательной реакции первого порядка при k1 k2 (k1 = 0,1c−1; k2 = 5,0 c−1; c01 =1,0 моль/ м3 ).
Штриховая кривая отвечает концентрации промежуточного вещества для стационарного режима
Режим протекания реакции, изображенный на рис. 2.12, называется квазистационарным и будет весьма подробно рассмотрен в 2.2.1.5.В, однако до перехода к данному вопросу остановимся еще на двух моментах: 1. вернемся еще раз к промежуточному веществу и 2. сформулируем некоторые соображения для последовательных реакций с k1 < k2 , т.е. для реакций с относительно неус-
тойчивым промежуточным веществом.
51
Х и м и ч е с к а я к и н е т и к а
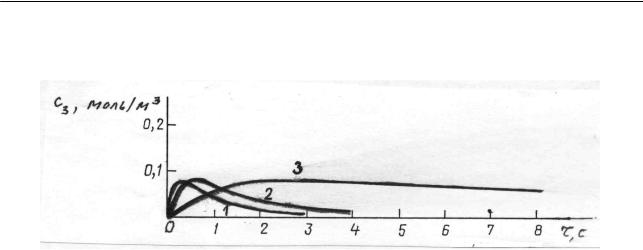
На рис. 2.13 приведены три кривые изменения концентрации промежуточного вещества, когда последнее относительно мало устойчиво – константа его разложения k2 в 10 раз превышает константу образования k1 . Высота максиму-
Рис. 2.13. Изменение концентрации промежуточного вещества при отношении констант k2 / k1=10:
1 − k1 =1,0 c−1, k2 =10,0 с−1; 2 − k1 = 0,5 c−1, k2 = 5,0 c−1; 3 − k1 = 0,1 c−1, k2 =1,0 c−1; c01 =1,0 моль/ м3
ма, являющаяся функцией γ cм. уравнение |
(2.133) во всех случаях одинакова, |
||||||
|
|
|
|
|
|
|
|
но вид кривых различен, так как время |
|
достижения |
максимума, согласно |
||||
(2.132), которое путем подстановки величины γ можно переписать в виде |
|||||||
|
ln |
k2 |
|
|
|
||
|
k |
(2.153) |
|||||
|
|
|
|||||
τmax = |
|
|
|
1 |
, |
||
k2 |
|
|
|
||||
|
− k1 |
|
|||||
зависит также от разности констант. Особый интерес представляет кривая 3, показывающая, что уменьшение разности k2 − k1 при том же их отношении
приводит к растягиванию максимума во времени. Концентрация промежуточного вещества длительное время держится приблизительно на одном низком уровне, близком к максимальному. Это дает право говорить о том, что если k1 k2 , то по истечении некоторого малого времени после начала реакции ус-
танавливается приблизительно стационарная (квазистационарная) концен-
трация неустойчивого промежуточного вещества, которая сохраняется в течение значительной части всего времени хода реакции. Рассмотренные результаты коррелируют с приведенными выше выводами.
В случае k1 < k2 , т.е. в случае, когда промежуточное вещество относительно неустойчиво, через достаточно большой промежуток времени член e−k1τ в уравнении (2.130) станет много больше члена e−k2τ . Поэтому уравнение (2.130) примет вид
52

Х и м и ч е с к а я к и н е т и к а
c |
= |
|
c0k1 |
e−k1τ . |
(2.154) |
||||
|
|||||||||
3 |
|
|
k2 − k1 |
|
|
|
|||
|
|
|
|
|
|
||||
Разделив (2.154) на (2.129) получим соотношение |
|
||||||||
|
c3 |
|
= |
|
|
k1 |
|
, |
(2.155) |
|
c |
k |
2 |
− k |
|||||
|
|
|
|
||||||
|
1 |
|
|
|
|
1 |
|
|
|
выражающее постоянство отношения концентраций промежуточного и исходного веществ в стационарном состоянии. Иногда такое состояние называют
переходным равновесием.
Как уже было показано см. уравнение(2.152) , к несколько иным результатам приходим в случае, когда k1 не просто меньше k2 , но и по абсолютной величине очень мало, т.е. k1 k2 . Уравнение (2.152), с учетом (2.39)и, исходя
из того, что средняя продолжительность жизни вещества при его распаде
τl =1/ k , можно представить в виде
c |
|
k |
|
τ(P) |
|
τ(P) |
|
(2.156) |
3 |
= |
1 |
= |
l |
= |
1/ 2 |
, |
|
|
|
|||||||
c1 |
|
k2 |
|
τl(A) |
|
τ1/(A2) |
|
|
где τl(P), τ1/(P2) и τl(A), τ1/(A2) – средняя продолжительность жизни и период полу-
превращения (полураспада) промежуточного и исходного веществ соответственно.
Таким образом, в стационарном состоянии концентрации промежуточ-
ного и исходного веществ будут относиться друг к другу как их средние продолжительности жизни или периоды полупревращения (полураспада).
Иногда в таких случаях говорят о вековом равновесии.
В. Квазистационарное и квазиравновесное приближения Из приведенных в 2.2.1.5.А решений прямой и обратной кинетических за-
дач следует, что даже в случае простейшей двустадийной односторонней мономолекулярной реакции, протекающей в закрытой системе, для кинетического описания приходится составлять и решать систему дифференциальных уравнений. Для более сложных реакций получить решение в аналитическом виде часто вообще не удается. В то же время, для определенных видов реакций и в определенных условиях процесс может протекать в стационарном или квазистационарном режиме, что существенно упрощает кинетические расчеты.
Стационарный режим протекания процесса может установиться в открытой системе. По смыслу стационарный режим напоминает ситуацию, которая устанавливается в ванне, если в нее с постоянной скоростью льется вода из крана и
стой же скоростью вытекает вода через нижний слив. При протекании реакции
встационарном режиме концентрации всех промежуточных веществ в данной точке пространства остаются постоянными, а, следовательно, в течение всего
53

Х и м и ч е с к а я к и н е т и к а
времени протекания процесса в стационарном режиме скорости образования и расходования всех промежуточных веществ одинаковы:
c |
= const; |
dci |
= 0, |
(2.157) |
|
||||
i |
|
dτ |
|
|
|
|
|
||
где ci - концентрация i − го промежуточного соединения в момент времени τ .
Для быстрого установления и последующего сохранения стационарного режима протекания реакции промежуточные вещества должны быть достаточно активными (продолжительность их жизни должна быть небольшой по сравнению со временем протекания реакции) и их концентрация должна быть достаточна мала. Из уже рассмотренного нами принципа независимости, справедливого для реакций, протекающих как в закрытых, так и в открытых системах,
вытекает важное следствие. Если в системе протекает несколько элементар-
ных реакций или стадий одной сложной реакции с участием одного и того же вещества, то изменение концентрации последнего будет равно алгебраической сумме скоростей каждой стадии, умноженных на стехиометрический коэффициент этого вещества в данной стадии. Именно указанная ал-
гебраическая сумма и приравнивается к нулю в уравнении (2.157). В результа-
те получается система алгебраических уравнений, позволяющая выразить концентрации промежуточных частиц, а затем и скорость реакции, через концентрации исходных веществ.
Метод расчета с использованием условия (2.157) при постоянстве концен-
трации исходных веществ называется методом стационарных концентраций
Боденштейна.
Опуская выводы, отметим, например, что для двустадийной реакции (2.126), состоящей из односторонних элементарных стадий первого порядка,
протекающей в открытой системе, после установления стационарного режима справедливо
cP |
=1 − e−k2τ , |
(2.158) |
|
||
сP(ст) |
|
|
где cP(ст) – стационарная концентрация промежуточного вещества P. Следовательно, формально стационарный режим, т.е. равенство cP =cP(ст), достигается
только через бесконечно большой промежуток времени: при τ = ∞ e−k2τ = 0 |
и |
||
|
cP |
=1. В то же время, достаточно малые, но конечные отклонения cP |
от |
|
cP(ст) |
||
|
|
|
|
cP(ст) |
достижимы за вполне приемлемое время. Так, если считать близкой к |
||
стационарной концентрацию сP =0,95cP(ст), то из (2.158) имеем
54

Х и м и ч е с к а я к и н е т и к а
|
|
1 |
|
|
|
c |
P |
|
|
3 |
|
(2.159) |
||
τ0,95 |
= |
|
|
ln 1 |
− |
|
|
|
= |
|
|
. |
||
k |
|
c |
|
|
k |
|
||||||||
|
|
2 |
|
|
P( |
|
|
|
2 |
|
|
|||
|
|
|
|
|
|
ст) |
|
|
|
|
||||
Например, для k2 =103 c−1 получаем малое время достижения стационарного
состояния – τ0,95 =3 10−3 c.
В закрытых системах концентрации исходных веществ уменьшаются во времени, а, следовательно, при протекании многостадийных реакций, например, реакции (2.126), концентрации промежуточных веществ не могут оста-
ваться постоянными. Для закрытых систем полезно понятие квазистационар-
ного режима реакции. Это режим, при котором концентрации промежуточ-
ных веществ в ходе процесса в каждый момент времени отвечают условиям стационарности по отношению к изменяющимся концентрациям исходных веществ, т.е. отношение концентраций промежуточных и исходных веществ остается постоянным. Таким образом, для квазистационарного режима реакции условие (2.157) остается в силе.
Подчеркнем два важных условия, при которых концентрации промежуточных частиц смогут «подстраиваться» к меняющейся концентрации исходного вещества в каждый момент времени и принимать значения, требуемые при стационарном протекании процесса.
Во-первых, время достижения квазистационарного режима должно быть коротким по сравнению со временем развития процесса в целом. Это время определяется константой скорости дальнейшего превращения промежуточной частицы. Значит, эта частица должна быть очень активна в своем дальнейшем превращении (или дальнейших превращениях) Следовательно, условие квазистационарности может быть применено только к активным промежуточным частицам. На примере рассматриваемой реакции (2.126) ясно, что данное усло-
вие достижимо при больших значениях k2 (k1 k2 ).
Во-вторых, предполагается, что максимальная концентрация промежуточных частиц достигается к моменту, когда концентрация исходных веществ практически не изменилась. Однако промежуточные частицы образуются из исходных веществ, малому изменению концентрации которых может соответствовать лишь очень малая (меньшая, чем само изменение) квазистационарная концентрация промежуточных частиц. Следовательно, условие квазистационарности применимо только к частицам, которые накапливаются в концентрациях, пренебрежимо малых по сравнению с концентрациями исходных веществ. Значит, оно может быть использовано только при малых значениях
k1 [k1 k2 для рассматриваемой реакции(2.126) .
Все вышеотмеченное согласуется с материалом, приведенным в 2.2.1.5.A при рассмотрении последовательных реакций. Таким образом, можно еще раз
55

Х и м и ч е с к а я к и н е т и к а
отметить, что для модельной реакции (2.126) условие квазистационарности применимо лишь для случая, когда k1 k2 .
Кинетика реакции (2.126) уже была рассмотрена в 2.2.1.5.А. Понятно, что ни при каких соотношениях k1 и k2 процесс не может быть квазистационарным
в течение периода времени до максимума на кинетической кривой для промежуточного вещества P. После достижения максимума режим реакции для случая k1 k2 (рис. 2.12) приближается к квазистационарному. Покажем это.
Зависимости концентраций исходного вещества A и промежуточного продукта P от времени для реакции (2.126) имеют, соответственно, вид
|
|
c |
A |
= c |
|
e−k1τ |
; |
(2.160) |
|
|
|
|
0 A |
|
|
|
|||
cP |
= c0 A |
|
|
|
k1 |
(e−k1τ |
− e−k2τ ). |
(2.161) |
|
k |
2 |
− k |
|||||||
|
|
|
|
1 |
|
|
|
|
|
Условие квазистационарности в рассматриваемом случае запишется в виде
dcP |
= k c |
A |
− k |
2 |
c |
P(ст) |
= 0; |
c |
P(ст) |
= |
k1 |
c |
A |
, |
(2.162) |
|
|
||||||||||||||
dτ |
1 |
|
|
|
|
|
k2 |
|
|
|
|||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
где cA – концентрация вещества A в данный конкретный момент времени τ .
Подставляя значение cA из (2.160) |
в |
(2.162), находим |
||||||||||||||||
|
|
|
c |
P( |
ст) |
= c |
|
k1 |
e−k1τ . |
(2.163) |
||||||||
|
|
|
|
|
||||||||||||||
|
|
|
|
|
|
|
|
0 A |
|
|
|
|||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
k2 |
|
|
|
|
Разделив почленно (2.161) |
на (2.163), имеем |
|
||||||||||||||||
|
|
c |
P |
|
|
= |
|
|
k |
2 |
|
|
[1 −e |
( 2 |
1) . |
(2.164) |
||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
− k |
−k τ |
|
||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||
|
|
cP(ст) |
|
|
|
|
k2 − k1 |
|
|
|
|
|
|
|||||
Анализ выражения (2.164) |
|
подтверждает тот факт, что квазистационарный |
||||||||||||||||
режим может |
быть реализован только при k1 k2 , |
когда можно пренебречь |
||||||||||||||||
величиной k1 |
в разности k2 − k1 и (2.164) переходит в уравнение (2.158) для |
|||||||||||||||||
стационарного процесса. Метод расчета, основанный на использовании условия (2.157) для промежуточных веществ при протекании реакции в закрытой сис-
теме, называется методом квазистационарных концентраций.
Если механизм протекания реакции по стадиям установлен, но неизвестны константы скорости отдельных стадий при условии, что промежуточные вещества достаточно активны, то часто для упрощения расчетов в качестве приближенной модели данной реакции принимают, что реакция протекает в квазистационарном режиме. При этом метод квазистационарных концентраций рас-
56

Х и м и ч е с к а я к и н е т и к а
сматривается как приближенный метод расчета, т.е. говорят о квазистацио-
нарном приближении.
Частным случаем квазистационарного приближения является квазиравновесное приближение. Оно наблюдается, если одна из стадий процесса обратима, причем равновесие устанавливается чрезвычайно быстро. Тогда можно считать, что на протяжении всей реакции между компонентами этой стадии сохраняется равновесие, т.е. они присутствуют в равновесных концентрациях.
Во многих случаях использование квазистационарного и квазиравновесного приближений приводит к кинетическим уравнениям, которые удовлетворительно описывают закономерности реального процесса.
При рассмотрении случаев несовпадения молекулярности и порядка реакции (см. 2.2.1.2) мы останавливались на вопросах, связанных с мономолекулярным распадом на примере диссоциации ацетона, сделав при этом ряд выводов, основываясь лишь на логических умозаключениях. Приведенный выше материал позволяет детализировать ряд вопросов, связанных с мономолекулярным распадом, с использованием кинетических закономерностей процесса. При этом, как и ранее, мы будем базироваться на считающейся общепринятой схеме Линдемана1, согласно которой активация молекул происходит путем двойного соударения, т.е. бимолекулярно.
Рассмотрим реакцию A → B + C, протекающую предположительно мономолекулярно. Согласно Линдеману механизм этой реакции включает следующие стадии:
(1) |
A + A → A + A |
k |
активационный процесс |
|
|
1 |
|
(2) |
A → B + C |
k2 |
распад |
(3) |
A + A → A + A |
k3 |
дезактивация |
В первом процессе появляется активированная молекула: возможно, относительная кинетическая энергия двух сталкивающихся молекул превращается в
колебательную энергию активированной молекулы A . Во втором процессе активированная молекула распадается. Процесс (2), однако, осуществляется не сразу после процесса (1), а по истечении некоторого конечного промежутка времени (в чем и состоит основное отличие от бимолекулярной реакции). Данный промежуток времени необходим для перераспределения энергии внутри молекулы и сосредоточения ее на связи, подлежащей разрыву. Так, например, для осуществления процесса (2) –- распада этана на два метильных радикала:
1Рассмотрение вопросов неудовлетворительности схемы Линдемана в целом, а также освещение других теорий мономолекулярных реакций выходят за рамки рассматриваемого материала. Эти вопросы достаточно подробно рассмотрены в [9]. Целью приведенного анализа являетсь лишь демонстрация использования принципа стационарности и более строгое, чем в 2.2.1.2, обоснование возможности протекания мономолекулярных реакций по различному порядку.
57

Х и м и ч е с к а я к и н е т и к а
H3C − CH3 → H3C + CH3 ,
по-видимому, необходимо сосредоточение энергии (возбуждения колебания) на связи C − C . При соударении же двух молекул в первую очередь, вероятно, возбуждаются колебания по периферическим связям C − H. Для перехода в необходимом количестве энергии с этих связей на связь C − C необходимо время. Пока же данный переход не осуществился, может произойти третье превращение – дезактивация, т.е. процесс обратный первому. При кинетическом расчете
активированная молекула |
A считается неустойчивым промежуточным про- |
|||||||||||||||
дуктом, к которому можно применить принцип стационарности: |
|
|||||||||||||||
|
dc |
A |
|
= k c2 |
− k |
c |
|
− k c |
|
c |
|
= 0 . |
(2.165) |
|||
|
|
|
A |
A |
|
|||||||||||
|
dτ |
|
|
|||||||||||||
|
|
1 |
A |
|
2 |
|
3 |
|
|
A |
|
|
||||
Отсюда концентрация активированных молекул |
|
|
|
|
||||||||||||
|
|
|
|
|
|
|
|
|
k c2 |
|
|
|
|
(2.166) |
||
|
|
|
|
c |
|
= |
|
|
1 |
A |
. |
|
|
|
||
|
|
|
|
|
k2 |
|
|
|
|
|
||||||
|
|
|
|
|
A |
|
+ k3cA |
|
|
|
|
|
||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Наблюдаемая скорость распада A равна, очевидно, скорости второй реакции, т.е.
|
|
|
− |
dcA |
= k2c |
|
. |
|
(2.167) |
||
|
|
|
dτ |
A |
|
||||||
|
|
|
|
|
|
|
|
|
|
||
Подставляя в (2.167) выражение для концентрации A |
из формулы (2.166), |
||||||||||
получим выражение для скорости |
|
|
|
|
|
|
|
||||
|
dcA |
|
2 |
|
|
k1k2cA |
|
|
|||
− |
= |
k1k2cA |
|
= |
|
cA , |
(2.168) |
||||
|
|
|
|
|
|||||||
|
dτ |
k2 + k3cA k2 + k3cA |
|
||||||||
являющеeся итогом рассуждений Линдемана. Согласно (2.168) в целом поря-
док реакции распада A промежуточный между первым и вторым. Однако необходимо рассмотреть крайние случаи, а именно:
1. Концентрация (давление) A велика. В этом случае скорость дезактивации k3cA cA может оказаться много больше скорости распада k2cA , или k3cA k2 ,
и первым слагаемым в знаменателе (2.168) можно пренебречь. В результате получится кинетическое уравнение реакции первого порядка:
− |
dcA. |
= |
k1k2 |
cA = k(I )cA. |
(2.169) |
dτ |
|
||||
|
|
k3 |
|
||
58

Х и м и ч е с к а я к и н е т и к а
В этих условиях, очевидно, поддерживается статистически равновесная концентрация активированных молекул, и реакция происходит в соответствии с законом спонтанного разложения1.
2. Концентрация (давление) A мала. В этом случае скорость реакции (2) k2cA может оказаться много больше скорости дезактивации k3cA cA , или
k2 k3cA . Пренебрегая в данном случае в знаменателе (2.168) вторым слагаемым, получим кинетическое уравнение реакции второго порядка:
− |
dcA |
= k c2 |
, |
(2.170) |
|
|
|||||
|
dτ |
1 |
A |
|
|
|
|
|
|
|
|
появляющееся, очевидно, в связи с относительно большими промежутками времени между соударениями, в течение которых в пределе все активированные молекулы успевают прореагировать. В данном случае скорость наблюдаемой реакции равна скорости процесса активации (1). Следовательно, согласно схеме Линдемана [уравнению (2.168)], константа скорости2, вычисляемая по
первому порядку, вообще говоря, имеет вид
k |
I |
) |
= |
k1k2cA |
. |
(2.171) |
|
||||||
( |
|
|
k2 + k3cA |
|
||
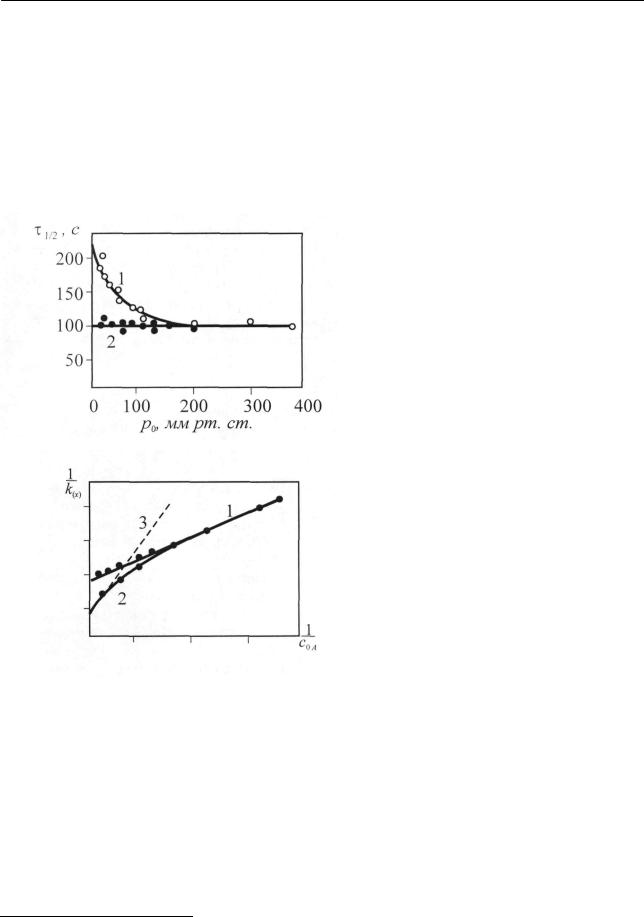
Следовательно, и «константа» первого порядка, и время превращения определенной доли вещества должны зависеть от концентрации или давления A. Как видно из рис. 2.14, при высоких давлениях период полураспада диэтилового эфира сохраняет постоянство, что свидетельствует о первом порядке реакции (см. 2.2.1.3), и увеличивается (реакция протекает уже не по первому порядку) с понижением давления ниже 200 мм рт. ст.3 Эту же зависимость можно пред-
ставить и в более удобной для обсуждения форме. Величина, обратная константе,
1 |
= |
k3 |
|
+ |
1 |
1 |
(2.172) |
|||
k |
(I ) |
k k |
2 |
k |
|
c |
A |
|||
|
|
|
|
|||||||
|
|
1 |
|
1 |
|
|
|
|||
1 Чисто статистическое рассмотрение мономолекулярного распада, выходящее за рамки нашего изложения, ведет к уравнению реакции первого порядка, т.е. к экспоненциальному закону. Единственным спорным допущением при этом является принятие распада молекулы событием индивидуальным, т.е. не зависящим от распада других молекул [9].
2В данном случае, как и при рассмотрении сложной реакции, состоящей из двух последовательных двусторонних элементарных стадий первого порядка (см. 2.2.1.5.А), речь идет не о константе скорости сложной реакции в целом, которой принципиально не существует, а об эффективной величине сложным образом зависящей от констант скорости всех стадий, а в некоторых случаях и от концентрации компонентов.
31 мм рт. ст. =1,33 Па [16].
59
Х и м и ч е с к а я к и н е т и к а
должна бы являться линейной функцией обратной концентрации. На рис. 2.15 представлены в спрямленном виде данные рис. 2.141. Видно, что ближе к началу координат наблюдается разброс точек, но все же можно провести прямую. Однако в большинстве исследованных таким образом реакций прямая не получается. Примерный ход экспериментальной кривой показан на рис. 2.15 кривой 2. В целом следует говорить о качественном подтверждении теории: при повышении давления константа скорости первого порядка стремится к постоянному значению (k∞ ). Таким образом, для каждой реакции существует минимальное
начальное давление, ниже которого константа первого порядка уменьшается.
Рис. 2.14. Зависимость периода полураспада диэтилового эфира от начального давления (p0 ) эфира:
1 – без водорода, 2 – с добавлением водорода
Рис. 2.15. Константа скорости мономолекулярной реакции в координатах 1/ k(I ) −1/ c0 A :
1 – диэтиловый эфир, 2 – обычное отклонение от прямолинейной зависимости, 3 – прямая, соответствующая уравнению (2.171)
Ранее (2.2.1.2) нами уже рассматривалось влияние примесей на порядок мономолекулярной реакции. При анализе кинетики мономолекулярных реакций всегда возникает вопрос о роли продуктов реакции и добавок посторонних инертных газов. Так, при разложении диэтилового эфира, добавление водорода к исходному эфиру препятствует уменьшению константы, поддерживая ее на
уровне k∞ (рис. 2.14, кривая 2). Эти результаты могут быть объяснены участи-
ем в активационном процессе (1) механизма Линдемана и продуктов реакции и заранее добавленного водорода. Подобное же компенсирующее свойство обнаруживают и другие вещества.
1 Рис. 2.14 и 2.15 приведены по [9] без изменений
60