

Деформация стеклообразных полимеров и явление вынужденной эластичности
Для стеклообразного состояния характерна упругая деформация:
![]() , (5)
, (5)
где ε – модуль упругости (модуль Юнга); σ – механическое напряжение (измеряется на динамометре); D – относительная деформация (удлинение).
При больших напряжениях в определенном температурном интервале стеклообразные полимеры способны подвергаться значительным деформациям – до нескольких сотен процентов. Такие деформации близки по своей природе к высокоэластическим, поэтому они были названы высокоэластическими (А.П. Александров, 1944 г.), а само явление – явлением вынужденной эластичности.
З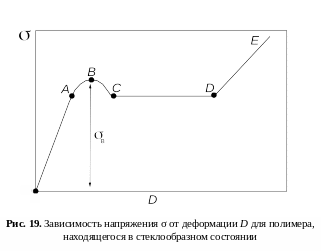 ависимость
напряжения σ
от деформации D
показана на рис. 19. Кривую можно разделить
на несколько участков.
ависимость
напряжения σ
от деформации D
показана на рис. 19. Кривую можно разделить
на несколько участков.
На участке oA соблюдается линейная зависимость напряжения от деформации, что соответствует обратимой упругой деформации.
На участке АВ тангенс угла наклона кривой к оси абсцисс с увеличением напряжения уменьшается. Это связано с началом развития в образце вынужденной эластичности. Под действием значительных внешних напряжений макромолекулы разворачиваются и ориентируются в направлении действия силы. В точке B (максимум на кривой) напряжение достигает значения, соответствующего пределу вынужденной эластичности σв.
На участке BC наблюдается спад напряжения. Это происходит из-за начала образования шейки. В точке С процесс формирования шейки заканчивается.
На отрезке СD происходит удлинение шейки за счет соседних, мало деформированных участков образца. На этом этапе σ = соnst, поэтому прямая СD параллельна оси абсцисс. В точке D весь образец переходит в шейку, т.е. его толщина становится равной толщине шейки. Рост шейки прекращается.
В области DE происходит дальнейшее удлинение образца как единого целого. Здесь, как и на первой стадии оА, зависимость напряжения от деформации линейна и подчиняется закону Гука.
У некоторых полимеров (например, нитрат целлюлозы) высокоэластическая деформация не сопровождается образованием шейки, в этом случае на деформационной кривой отсутствует максимум.
Вынужденная эластичность, как и высокоэластичность, носит релаксационный характер, т.е. зависит от времени и скорости деформации. Чем меньше время воздействия и больше скорость деформации, тем больше напряжение, а значит, выше значение предела вынужденной эластичности σв. При этом область вынужденной эластичности сокращается вплоть до исчезновения, когда образец разрушается ниже σв.
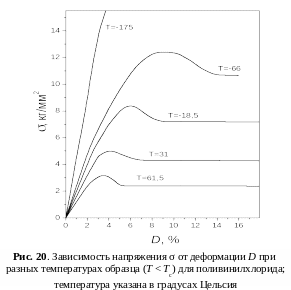
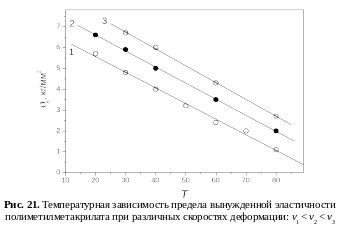
Аналогичная зависимость наблюдается с понижением температуры (рис. 20), что отвечает принципу температурно-временной суперпозиции: увеличение частоты действия силы эквивалентно понижению температуры (рис. 21). С повышением температуры уменьшается значение σв , что видно из рис. 20. При Т = Тс предел вынужденной эластичности становится равным нулю: σв = 0. При понижении температуры до значения, когда она становится равной температуре хрупкости Т = Тхр, напряжение, необходимое для перегруппировки сегментов под действием силы, достигает значения разрушающего напряжения σхр (σв = σхр). При этом происходит хрупкое разрушение материала.
Таким образом, температуру хрупкости можно определить как температуру, ниже которой полимер не проявляет вынужденной эластичности. Графически Тхр находится, как точка пересечения кривых зависимостей σхр = f(Т) и σв = f(Т) (рис. 22).
Температура хрупкости, в отличие от температуры стеклования, зависит не только от скорости воздействия силы, но и от вида деформации (сжатие, растяжение, сдвиг).
Итак, температурный интервал вынужденной эластичности снизу ограничен температурой хрупкости, а сверху – температурой стеклования. Чем больше разница между значениями Тс и Тхр, тем шире область вынужденной эластичности.
Т емпература
стеклования определяет верхний предел,
а температура хрупкости – нижний предел
температурной области эксплуатации
пластмасс, поэтому большой интервал
Тхр
–
Тс
– ценное свойство полимера.
емпература
стеклования определяет верхний предел,
а температура хрупкости – нижний предел
температурной области эксплуатации
пластмасс, поэтому большой интервал
Тхр
–
Тс
– ценное свойство полимера.
На характер температурной зависимости предела вынужденной эластичности от температуры оказывает влияние природа полимера: наличие полярных групп, плотность упаковки и молекулярная масса.
С усилением межмолекулярного взаимодействия вследствие присутствия полярных групп температурный интервал вынужденной эластичности расширяется.
При понижении плотности упаковки снижается температура хрупкости, что также приводит к расширению интервала Тхр – Тс.
С увеличением молекулярной массы температура стеклования повышается до степени полимеризации n ≈ 200, а температура хрупкости – до n ≈ 600. При n > 200 температура стеклования становится постоянной, а температура хрупкости понижается, т.к. величина разрушающего напряжения σхр продолжает увеличиваться. При n > 600 температура хрупкости сохраняет постоянное значение.
Изменение температур стеклования Тс, текучести Тт и хрупкости Тхр от молекулярной массы полимера показано на рис. 23.
