


Regions of the lung with a high  ratio and a high PO2 cannot compensate for regions with a low
ratio and a high PO2 cannot compensate for regions with a low  ratio and low PO2.
ratio and low PO2.
In marked contrast, a high PO2 in blood coming from a region with a high  ratio cannot fully
ratio cannot fully
compensate for blood with a low PO2 from a region with a low  ratio. The difference stems from the shape of the oxyhemoglobin dissociation curve, which is curvilinear (rather than linear) and becomes nearly flat at the top (see Fig. 1.5). After hemoglobin is nearly saturated with O2 (on the relatively flat part of the oxyhemoglobin dissociation curve), increasing PO2 only contributes to the very small amount of oxygen dissolved in blood and does not significantly boost the O2 content. In other words, because most of the oxygen content in blood is due to O2 bound to hemoglobin, once the hemoglobin is fully saturated, blood with a higher than normal PO2 does not have a correspondingly higher O2 content and cannot compensate for blood with a low PO2 and low O2 content.
ratio. The difference stems from the shape of the oxyhemoglobin dissociation curve, which is curvilinear (rather than linear) and becomes nearly flat at the top (see Fig. 1.5). After hemoglobin is nearly saturated with O2 (on the relatively flat part of the oxyhemoglobin dissociation curve), increasing PO2 only contributes to the very small amount of oxygen dissolved in blood and does not significantly boost the O2 content. In other words, because most of the oxygen content in blood is due to O2 bound to hemoglobin, once the hemoglobin is fully saturated, blood with a higher than normal PO2 does not have a correspondingly higher O2 content and cannot compensate for blood with a low PO2 and low O2 content.
In the normal lung, regional differences in the  ratio affect gas tensions in blood coming from specific regions, as well as gas tensions in the resulting arterial blood. At the apices, where the
ratio affect gas tensions in blood coming from specific regions, as well as gas tensions in the resulting arterial blood. At the apices, where the  ratio is approximately 3.3, PO2 = 132 mm Hg and PCO2 = 28 mm Hg. At the bases, where the
ratio is approximately 3.3, PO2 = 132 mm Hg and PCO2 = 28 mm Hg. At the bases, where the  ratio is approximately 0.63, PO2 = 89 mm Hg and PCO2 = 42 mm Hg. As discussed, the net PO2 and PCO2 of the combined blood coming from the apices, bases, and the areas between are a function of the relative amounts of blood from each of these areas and the gas contents of each.
ratio is approximately 0.63, PO2 = 89 mm Hg and PCO2 = 42 mm Hg. As discussed, the net PO2 and PCO2 of the combined blood coming from the apices, bases, and the areas between are a function of the relative amounts of blood from each of these areas and the gas contents of each.
In disease states, ventilation–perfusion mismatch frequently is much more extreme, resulting in clinically significant gas exchange abnormalities. When an area of lung behaves as a shunt or even as a
region having a very low  ratio, blood coming from this area has a low O2 content and saturation, which cannot be compensated for by blood from relatively preserved regions of lung.
ratio, blood coming from this area has a low O2 content and saturation, which cannot be compensated for by blood from relatively preserved regions of lung.  mismatch that is severe, particularly with areas of a high
mismatch that is severe, particularly with areas of a high  ratio, can effectively produce dead space and therefore decrease the
ratio, can effectively produce dead space and therefore decrease the  to other areas of the lung carrying a disproportionate share of the perfusion. Because CO2 excretion depends on
to other areas of the lung carrying a disproportionate share of the perfusion. Because CO2 excretion depends on  , PCO2 may rise unless an overall increase in the
, PCO2 may rise unless an overall increase in the 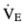 restores the effective
restores the effective  .
.
Abnormalities in gas exchange
The net effect of disturbances in the normal pattern of gas exchange can be assessed by measurement of the gas tensions (PO2 and PCO2) in arterial blood. The information that can be obtained from arterial blood gas measurement is discussed further in Chapter 3, but the mechanisms of hypoxemia (decreased arterial PO2) and hypercapnia (increased PCO2) are considered here because they relate to the physiologic principles just discussed.
Hypoxemia
Blood that has traversed pulmonary capillaries leaves with a PO2 that should be in equilibrium with and almost identical to the PO2 in companion alveoli. Although it is difficult to measure the O2 tension in alveolar gas, it can be conveniently calculated by a formula known as the alveolar gas equation. A

simplified version of this formula is relatively easy to use and can be extremely useful in the clinical setting, particularly when trying to deduce why a patient is hypoxemic. The alveolar O2 tension (PAO2)c can be calculated by Eq. 1.8:
where FIO2 = fractional content of inspired O2 (FIO2 of air = 0.21), PB = barometric pressure (approximately 760 mm Hg at sea level), PH2O = vapor pressure of water in the alveoli (at full saturation at 37°C, PH2O = 47 mm Hg), PACO2 = alveolar CO2 tension (which can be assumed to be identical to arterial CO2 tension, PaCO2), and R = respiratory quotient (CO2 production divided by O2 consumption, usually approximately 0.8). In practice, for the patient breathing room air (FIO2 = 0.21), the equation often is simplified. When numbers are substituted for FIO2, PB, and PH2O and when PaCO2 is used instead of PACO2, the resulting equation (at sea level) is Eq. 1.9:
The simplified alveolar gas equation (see Eq. 1.9) can be used to calculate alveolar PO2 (PAO2) for the patient breathing room air.
By calculating PAO2, the expected PaO2 can be determined. Even in a normal person, PAO2 is greater than PaO2 by an amount called the alveolar-arterial oxygen difference or gradient (AaDO2). A gradient exists even in normal individuals for two main reasons: (1) A small amount of cardiac output behaves as a shunt, without ever going through the pulmonary capillary bed. This includes venous blood from the bronchial circulation, a portion of which drains into the pulmonary veins, and coronary venous blood draining via thebesian veins directly into the left side of the heart. Desaturated blood from these sources lowers O2 tension in the resulting arterial blood. (2) Ventilation–perfusion gradients from the top to the bottom of the lung result in somewhat less oxygenated blood from the bases combining with better oxygenated blood from the apices.
AaDO2 normally is less than 15 mm Hg, but it increases with age. AaDO2 may be elevated in disease for several reasons. First, a shunt may be present so that some desaturated blood combines with fully saturated blood and lowers PO2 in the resulting arterial blood. Common causes of a shunt are as follows:
1.Intracardiac lesions with a right-to-left shunt at the atrial or ventricular level (e.g., an atrial or ventricular septal defect). Note that although a left-to-right shunt can produce severe long-term cardiac consequences, it does not affect either AaDO2 or arterial PO2 because its net effect is to
recycle already oxygenated blood through the pulmonary vasculature, not to dilute oxygenated blood with desaturated blood.
2.Structural abnormalities of the pulmonary vasculature that result in direct communication between pulmonary arterial and venous systems (e.g., pulmonary arteriovenous malformations).
3.Pulmonary diseases that result in filling of the alveolar spaces with fluid (e.g., pulmonary edema)
or complete alveolar collapse. Either process can result in complete loss of ventilation to the affected alveoli, although some perfusion through the associated capillaries may continue.
Although perfusion of poorly ventilated lung units is detrimental to gas exchange, it can be helpful
Данная книга находится в списке для перевода на русский язык сайта https://meduniver.com/

for immunologic reasons, such as delivering white blood cells to an area of pneumonia.
Ventilation–perfusion mismatch and shunting are the two important mechanisms for elevation of AaDO2.
AaDo2, Alveolar-arterial O2 difference.
Another cause of elevated AaDO2 is ventilation–perfusion mismatch. Even when total ventilation and total perfusion to both lungs are normal, if some areas receive less ventilation and more perfusion (low  ratio) whereas others receive more ventilation and less perfusion (high
ratio) whereas others receive more ventilation and less perfusion (high  ratio), AaDO2 increases and hypoxemia results. As just mentioned, the reason for this phenomenon is that areas having a low
ratio), AaDO2 increases and hypoxemia results. As just mentioned, the reason for this phenomenon is that areas having a low  ratio provide relatively desaturated blood with a low O2 content. Blood coming from regions
ratio provide relatively desaturated blood with a low O2 content. Blood coming from regions
with a high  ratio cannot compensate for this problem because the hemoglobin is already fully saturated and cannot increase its O2 content further by increased ventilation (Fig. 1.9).
ratio cannot compensate for this problem because the hemoglobin is already fully saturated and cannot increase its O2 content further by increased ventilation (Fig. 1.9).

FIGURE 1.9 Example of nonuniform ventilation producing  mismatch in twoalveolus model. In this instance, perfusion is equally distributed between the two alveoli. Calculations demonstrate how
mismatch in twoalveolus model. In this instance, perfusion is equally distributed between the two alveoli. Calculations demonstrate how  mismatch lowers arterial PO2 and causes elevated alveolar-arterial oxygen difference. Source: (Modified from Comroe, J. H. (1962). The lung (2nd ed., p. 94). Chicago, IL: Year Book Medical Publishers.)
mismatch lowers arterial PO2 and causes elevated alveolar-arterial oxygen difference. Source: (Modified from Comroe, J. H. (1962). The lung (2nd ed., p. 94). Chicago, IL: Year Book Medical Publishers.)
In practice, the contribution to hypoxemia of true shunt ( 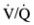 = 0) and
= 0) and  mismatch (with areas of
mismatch (with areas of  that are low but not 0) can be distinguished by having the patient inhale 100% O2. In the former case, increasing inspired PO2 does not add more O2 to the shunted blood and O2 content does not increase significantly. In the latter case, alveolar and capillary PO2 rise considerably with additional O2, fully
that are low but not 0) can be distinguished by having the patient inhale 100% O2. In the former case, increasing inspired PO2 does not add more O2 to the shunted blood and O2 content does not increase significantly. In the latter case, alveolar and capillary PO2 rise considerably with additional O2, fully
saturating blood coming even from regions with a low ratio, and arterial PO2 rises substantially.
Данная книга находится в списке для перевода на русский язык сайта https://meduniver.com/

A third cause of elevated AaDO2 occurs primarily in specialized circumstances. This cause is a “diffusion block” in which PO2 in pulmonary capillary blood does not reach equilibrium with alveolar gas. If the interface (i.e., the tissue within the alveolar wall) between the capillary and the alveolar lumen is thickened, one can hypothesize that O2 does not diffuse as readily and that the PO2 in pulmonary capillary blood never reaches the PO2 of alveolar gas. However, even with a thickened alveolar wall, there is still sufficient time for this equilibrium. Unless the transit time of erythrocytes through the lung is significantly shortened, failure to equilibrate does not appear to be a problem. A specialized circumstance, in which a diffusion block plus more rapid transit of erythrocytes together contribute to hypoxemia, occurs during exercise in a patient with interstitial lung disease, as will be discussed later. However, for most practical purposes in the nonexercising patient, a diffusion block should be considered only a hypothetical rather than a real mechanism for increasing AaDO2 and causing hypoxemia.
Increasing the difference between alveolar and arterial PO2 is not the only mechanism that results in hypoxemia. Alveolar PO2 can be decreased, which must necessarily lower arterial PO2 if AaDO2 remains constant. Referring to the alveolar gas equation, it is relatively easy to see that alveolar PO2 drops if barometric pressure falls (e.g., with altitude) or if alveolar PCO2 rises (e.g., with hypoventilation). In the
latter circumstance, when total  falls, PCO2 in alveolar gas rises at the same time alveolar PO2 falls. Hypoventilation is relatively common in lung disease and is defined by the presence of a high PCO2 accompanying the hypoxemia. If PCO2 is elevated and AaDO2 is normal, then hypoventilation is the
falls, PCO2 in alveolar gas rises at the same time alveolar PO2 falls. Hypoventilation is relatively common in lung disease and is defined by the presence of a high PCO2 accompanying the hypoxemia. If PCO2 is elevated and AaDO2 is normal, then hypoventilation is the
exclusive cause of low PO2. If AaDO2 is elevated, either  mismatch or shunting also contributes to hypoxemia.
mismatch or shunting also contributes to hypoxemia.
WHEN HYPOVENTILATION IS THE SOLE CAUSE OF HYPOXEMIA, AaDO 2 IS NORMAL
Mechanisms of hypoxemia:
1.Shunt
2. mismatch
mismatch
3.Hypoventilation
4.Low inspired PO2
In summary, lung disease can result in hypoxemia for multiple reasons. Shunting and ventilation– perfusion mismatch are associated with elevated AaDO2. They often can be distinguished, if necessary, by
inhalation of 100% O2, which markedly increases PaO2 with  mismatch but not with true shunting. In contrast, hypoventilation (identified by high PaCO2) and low inspired PO2 lower alveolar PO2 and cause hypoxemia, although AaDO2 remains normal. Because many of the disease processes examined in this text lead to several pathophysiologic abnormalities, it is not at all uncommon to see more than one of the aforementioned mechanisms producing hypoxemia in a particular patient.
mismatch but not with true shunting. In contrast, hypoventilation (identified by high PaCO2) and low inspired PO2 lower alveolar PO2 and cause hypoxemia, although AaDO2 remains normal. Because many of the disease processes examined in this text lead to several pathophysiologic abnormalities, it is not at all uncommon to see more than one of the aforementioned mechanisms producing hypoxemia in a particular patient.
Hypercapnia
As discussed earlier in the section on Ventilation,  is the prime determinant of arterial PCO2,
is the prime determinant of arterial PCO2,