


persons with the ZZ genotype (who are commonly said to have α1-antitrypsin deficiency because of low serum levels). As mentioned earlier, the structural integrity of alveolar walls appears to depend on the balance between elastin degradation by elastase and protection from this destruction afforded by α1- antitrypsin. In patients with α1-antitrypsin deficiency, lack of the elastase inhibitor is believed to permit elastase action to proceed in an unchecked fashion, and early development of emphysema is the consequence.
Another factor of interest, one that presumably is at least partially genetically determined, is the degree of the patient’s preexisting bronchial hyperresponsiveness. Data support the hypothesis that accelerated decline in lung function occurs in patients who have greater levels of bronchial responsiveness. However, this is an area of controversy, in part because the potential for smoking to induce changes in bronchial responsiveness makes it difficult to determine cause/effect relationships.
Pathology
Much of the pathology in chronic bronchitis relates to mucus and the mucus-secreting apparatus in the airways. Mucus-secreting glands and goblet cells are responsible for the production of bronchial secretions, but the mucous glands are the more important source (see Chapter 4). In chronic bronchitis, enlargement (hypertrophy) of the mucus-secreting glands has been objectively assessed by comparing the relative thickness of the mucous glands with the total thickness of the airway wall. This ratio, known as the Reid index, is increased in patients with chronic bronchitis. In general, the number of goblet cells in the airways is increased as well (hyperplasia), and these cells are abundant in airways more peripheral than normal. These alterations in the mucus-secreting apparatus increase the quantity of airway mucus, and its composition is likely altered as well. In practice, the secretions found in patients are often thick and more viscous than usual. Bronchial walls demonstrate evidence of an inflammatory process, with cellular infiltration and variable degrees of fibrosis.
Chronic bronchitis is characterized by enlargement of the mucus-secreting glands and an increased number of goblet cells.
In the smaller airways (e.g., bronchioles), inflammation, fibrosis, intraluminal mucus, and an increase in goblet cells all contribute to a decrease in luminal diameter. Because the resistance of airways varies inversely with the fourth power of the radius, even small changes in bronchiolar size may result in major impairment to airflow at the level of the small airways. These pathologic changes in the small airways are thought to be a major contributor to the airflow obstruction in COPD, particularly in patients with mild disease.
In patients with severe chronic airflow obstruction, the most important process responsible for airflow obstruction is emphysema. As mentioned earlier, the pathology of emphysema is characterized by the destruction of alveolar walls and the enlargement of terminal air spaces. Several types of emphysema have distinct pathologic features, which are primarily dependent on the distribution of the lesions. The most important types are panacinar (panlobular) emphysema and centriacinar (centrilobular) emphysema (Figs. 6.2 and 6.3). Panacinar emphysema is characterized by a relatively uniform involvement of the acinus, the region beyond the terminal bronchiole, including respiratory bronchioles, alveolar ducts, and alveolar sacs. Examination of a section of lung with panacinar emphysema shows that the damage in an involved area is relatively diffuse (Fig. 6.4). Typically, the lower zones of the lung are more involved than the upper zones. Panacinar emphysema is the usual type of emphysema described in patients who have α1-antitrypsin deficiency, although the condition is not limited to this clinical setting.
Данная книга находится в списке для перевода на русский язык сайта https://meduniver.com/

FIGURE 6.2 Diagrams of panlobular (A) and centrilobular (B) emphysema. In panlobular (panacinar) emphysema, enlargement of air spaces is relatively uniform throughout the acinus. In centrilobular (centriacinar) emphysema, the enlargement of air spaces is primarily at the level of respiratory bronchioles. A, alveolus; AD, alveolar duct; AS, alveolar sac; RB1, RB2, RB3, three generations of respiratory bronchioles; TB, terminal bronchiole. Source: (From Thurlbeck, W. M. (1968). Chronic obstructive lung disease. In S. C. Sommers (Ed.), Pathology annual (Vol 3). New York, NY: Appleton-Century-Crofts)
FIGURE 6.3 Low-power photomicrographs of emphysema. A, Centrilobular (centriacinar) emphysema with dilation of airspaces surrounding a bronchiole. B,
Panlobular (panacinar) emphysema with more diffuse airspace dilation.
Source: (From Leslie, K. O., & Wick, M. R. (2018). Practical pulmonary pathology. A diagnostic approach (3rd ed.). Philadelphia, PA: Elsevier.)
FIGURE 6.4 The mounted section of the whole lung shows diffuse involvement
seen with panacinar emphysema. Source: (From Thurlbeck, W. M. (1976). Chronic
airflow obstruction in lung disease. Philadelphia, PA: WB Saunders.)
Pathologic changes from smoking often start in small airways, predating the advanced findings associated with chronic bronchitis and emphysema.
In centrilobular emphysema, the predominant involvement and dilation are found in the proximal part of the acinus, the respiratory bronchiole. The appearance of a lung section with centrilobular emphysema is different from that with panacinar emphysema. In centrilobular emphysema, involvement in an affected area seems to be more irregular, with apparently spared alveolar tissue between the dilated respiratory bronchioles at the center of the acinus (Fig. 6.5). This type of emphysema is the typical form seen in smokers. It is reasonable to speculate that the prominent involvement focused around the respiratory bronchiole is a consequence of extension of the bronchiolar inflammation in mild COPD.
Данная книга находится в списке для перевода на русский язык сайта https://meduniver.com/

FIGURE 6.5 The mounted section of the whole lung shows centrilobular
emphysema. Adjacent to emphysematous spaces (which represent dilated
respiratory bronchioles) are spared areas of lung parenchyma (representing alveolar
ducts and alveolar spaces). Source: (From Thurlbeck, W. M. (1967). Internal
surface area and other measurements in emphysema. Thorax, 22, 483–496. BMJ
Publishing Group.)
Pathophysiology
Underlying a discussion of the pathophysiology of COPD is the fact that cigarette smoking affects the large airways, small airways, and pulmonary parenchyma. The pathophysiologic consequences resulting from disease at each of these levels contribute to the overall clinical picture of COPD. In addition, the degree of airway reactivity, which probably is affected by genetic and environmental factors, appears to modify the clinical expression of disease in a given patient. This section simplifies, summarizes, and places into a conceptual framework some of the information regarding structure–function correlations for each of these aspects of COPD.
Functional abnormalities in airways disease
In the larger airways (bronchi), an increase in the mucus-secreting apparatus and the amount of mucus

produced results in the symptoms of excessive cough and sputum production characteristic of chronic bronchitis. However, these symptoms do not necessarily correlate with the degree of airflow obstruction, as some patients with typical symptoms of chronic bronchitis do not exhibit abnormally high resistance or changes in other measurements of airflow. When airflow obstruction exists, in general, additional pathologic factors, either in the small airways (inflammation and fibrosis) or pulmonary parenchyma (emphysema), are critical for the presence of obstruction. In relatively mild airflow obstruction associated with chronic bronchitis, disease in the small airways is likely the important factor responsible for airflow obstruction. When airflow obstruction is more marked, coexisting emphysema, with decreased caliber of small airways due to loss of airway tethering, is often the primary reason for the obstruction.
Small airways disease, emphysema, or both contribute significantly to decreased expiratory flow rates in COPD.
In patients who have a component of airway hyperreactivity contributing to their disease, often the clinical expression is that of an asthma-COPD overlap syndrome. Airway smooth muscle constriction adds more reversible airflow obstruction than is typically seen in the patient without airway hyperreactivity.
The common problem produced by the processes affecting airways is a decrease in the overall crosssectional area of the airways. Airways resistance is increased (i.e., worsened) by anything that reduces the cross-sectional area of the lumen of the airways: intraluminal secretions, bronchospasm, or thickening of the airway wall caused by edema, inflammatory cells, fibrosis, or enlargement of the mucus-secreting apparatus, for example. When disease is located primarily in the peripheral airways and is mild, the functional consequences may be relatively subtle. Because the peripheral airways contribute only approximately 10% to 20% of overall airways resistance, total resistance is preserved unless small airways disease is considerable, or additional disease affects the larger airways.
As another consequence of airway disease, expiratory flow rates—including forced expiratory volume in 1 second (FEV1), FEV1/forced vital capacity (FVC) ratio, and maximal mid-expiratory flow rate (MMFR)—are generally decreased. The use of inhaled bronchodilators may or may not result in significantly improved flow rates in COPD. Patients with asthma-COPD overlap syndrome and greater airway reactivity generally have the most striking improvement in flow rates after receiving an inhaled bronchodilator.
Before a discussion of how lung volumes change in patients having the airway disease associated with COPD, it is useful to review the factors that determine major lung volumes: total lung capacity (TLC), functional residual capacity (FRC), and residual volume (RV). TLC is the point at which the maximal force of the inspiratory muscles acting to expand the lungs is equaled by the elastic recoil of the respiratory system (primarily lung recoil) resisting expansion (see Chapter 1). At FRC, the resting point of the respiratory system, there is a balance between the elastic recoil of the lungs and the elastic recoil of the chest wall, which are acting in opposite directions—the lungs inward and the chest wall outward. The determinants of RV depend to some extent on age. In a normal young person, RV is the point at which the relatively stiff chest wall can be compressed no further by the expiratory muscles. With increasing age, a sufficient number of airways close at low lung volumes to limit further expiration, and airway closure is an important determinant of RV. In disease states in which airways are likely to close at low lung volumes, airway closure is associated with an elevated RV, even in young patients.
In patients with pure airway disease, TLC theoretically remains close to normal because neither the elastic recoil of the lung nor inspiratory muscle strength is altered. Similarly, FRC should remain normal because the recoil of the lung and the recoil of the chest wall are unchanged. However, if expiratory flow
rates are decreased and the respiratory rate is high, the patient may not have sufficient time during
Данная книга находится в списке для перевода на русский язык сайта https://meduniver.com/

expiration to reach the normal resting end-expiratory point. When this occurs, the end-expiratory lung volume is increased, resulting in an increase in the measured FRC. RV is generally also increased with processes that involve airways, because the narrowing and occlusion of small airways by secretions and inflammation result in air trapping during expiration.
Functional abnormalities in emphysema
Although emphysema (i.e., destruction of alveolar walls) leads to decreased expiratory flow rates, the pathophysiology is different from the situation in pure airway disease. The primary problem in emphysema is loss of elastic recoil (i.e., loss of the lung’s natural tendency to resist and recover from expansion). An important consequence of decreased elastic recoil is a decreased driving pressure that expels air from the alveoli during expiration. A simple analogy is a balloon filled with air, in which the elastic recoil is the “stiffness” of the balloon. With a given volume of air inside an unsealed balloon, a stiffer balloon will expel air more rapidly than a less stiff balloon. An emphysematous lung is like a less stiff balloon: a smaller than normal force drives air out of the lungs during expiration.
In emphysema, decreased expiratory flow rates are largely due to loss of elastic recoil of the lung, resulting in:
1.Lower driving pressure for expiratory airflow
2.Loss of radial traction on the airways provided by supporting alveolar walls, thus promoting airway collapse during expiration
Loss of driving pressure during exhalation is not the only consequence of emphysema. There is also an indirect effect on the collapsibility of airways. Normally, the walls of airways are held open and pulled radially outward from the center of their lumen by a supporting structure of tissue from the adjacent lung parenchyma. When the alveolar tissue is disrupted, as in emphysema, the supporting structure for the airways is diminished, and less radial traction is exerted to prevent airway collapse (Fig. 6.6). During a forced expiration, the strongly positive pleural pressure promotes collapse. Airways lacking an adequate supporting structure are more likely to collapse (and have diminished flow rates and air trapping) than normally supported airways.

FIGURE 6.6 Schematic diagram of radial traction exerted by alveolar walls
(represented as springs), acting to keep the airways open. A, Normal situation. B,
Loss of radial traction as seen in emphysema.
The decrease in elastic recoil in emphysema also alters the compliance curve of the lung and measured lung volumes. The compliance curve relates transpulmonary pressure and the associated volume of gas within the lung (see Chapter 1). Because an emphysematous lung has less elastic recoil (i.e., is less stiff), it resists expansion less than its normal counterpart, the compliance curve is shifted upward and to the left, and the lung has more volume at any particular transpulmonary pressure (Fig. 6.7). TLC is increased because loss of elastic recoil results in a smaller force opposing the action of the inspiratory musculature. FRC is also increased because the balance between the outward recoil of the chest wall and the inward recoil of the lung is shifted in favor of the chest wall. As in bronchitis, RV is substantially increased in emphysema because poorly supported airways are more susceptible to closure during a maximal expiration.
Данная книга находится в списке для перевода на русский язык сайта https://meduniver.com/

FIGURE 6.7 Compliance curve of lung in emphysema compared with that of
normal lung. In addition to shift of curve upward and to left, total lung capacity in
emphysema (point B on volume axis) is greater than normal total lung capacity
(point A). In pure chronic bronchitis without emphysema, the compliance curve is
normal.
Mechanisms of abnormal gas exchange
In obstructive lung disease, many of the observed pathologic changes affecting airflow are not uniformly distributed. For example, in chronic bronchitis, some airways are extensively affected by secretions and plugging, but others remain relatively uninvolved, so ventilation is not uniformly distributed throughout the lung. Regions of the lung supplied by more diseased airways receive diminished ventilation in comparison with regions supplied by less diseased airways. Although there may be a compensatory decrease in blood flow to underventilated alveoli, the compensation is not totally effective, and inequalities and mismatching of ventilation and perfusion result. This type of ventilation-perfusion disturbance, with some areas of lung having low ventilation-perfusion ratios and contributing desaturated blood, leads to arterial hypoxemia.
In obstructive lung disease, nonuniformity of the disease process results in 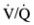 mismatch and hypoxemia.
mismatch and hypoxemia.
Carbon dioxide elimination is impaired in some patients with obstructive lung disease. The mechanism of alveolar hypoventilation and CO2 retention is less clear than the mechanism of hypoxemia. Several factors probably contribute, including increased work of breathing (due to impaired airflow),

abnormalities of central ventilatory drive, and ventilation-perfusion mismatch creating some areas with high ventilation-perfusion ratios that effectively act as dead space.
Mechanisms that contribute to alveolar hypoventilation and CO2 retention in obstructive lung disease are as follows:
1.Increased work of breathing
2.Abnormalities of ventilatory drive
3. mismatch
mismatch
4.Decreased effectiveness of the diaphragm
An additional problem—fatigue of inspiratory muscles—has received attention as a factor contributing to acute CO2 retention when affected patients are in respiratory failure (see Chapter 19). The importance of diaphragmatic fatigue in the stable patient with chronic hypercapnia is less certain. However, it is clear that contraction of the diaphragm, the major muscle of inspiration, is less efficient and less effective in patients with obstructive lung disease. When FRC is increased, the diaphragm is lower and flatter, and its fibers are shortened even before the initiation of inspiration. A shortened, flattened diaphragm is at a mechanical disadvantage compared with a longer, curved diaphragm and is less effective as an inspiratory muscle.
Pulmonary hypertension
A potential complication of COPD is the development of pulmonary hypertension (i.e., abnormally high pressures within the pulmonary arterial system). Long-standing pulmonary hypertension places an added workload onto the right ventricle, which hypertrophies and eventually may fail. The term cor pulmonale is used to describe the disease of the right ventricle secondary to lung disease (either COPD or other forms of lung disease); this topic is discussed in Chapter 14. The primary feature of COPD that leads to pulmonary hypertension and eventually to cor pulmonale, is chronic hypoxia. A decrease in PO2 is a strong stimulus to the constriction of pulmonary arterioles (see Chapter 12). If hypoxia is corrected, the element of pulmonary vasoconstriction may be reversible, but vascular remodeling from chronic hypoxia may not fully reverse.
The major cause of pulmonary hypertension in COPD is hypoxia. Additional factors include hypercapnia, polycythemia, and destruction of the pulmonary vascular bed.
Several other but relatively less important factors that may contribute to elevated pulmonary artery pressure are hypercapnia, polycythemia, and reduction in the area of the pulmonary vascular bed. Hypercapnia, like hypoxia, can cause pulmonary vasoconstriction. To a large extent, this effect may be mediated by the change in pH resulting from an increase in PCO2. An elevation in hematocrit (i.e., polycythemia) is often found in the chronically hypoxemic patient, producing increased blood viscosity and contributing to elevated pulmonary artery pressure. Finally, in emphysema, destruction of alveolar walls is accompanied by a loss of pulmonary capillaries. Therefore, in extensive disease, the limited pulmonary vascular bed may result in a high resistance to blood flow and, consequently, an increase in pulmonary artery pressure.
Данная книга находится в списке для перевода на русский язык сайта https://meduniver.com/