

4. Катализ [1, § 60; 2, § 6.2.6]
К изложенному в учебнике необходимо несколько уточнений и добавлений.
Во-первых, реакция в присутствии катализатора идёт не в одну стадию, а минимум в две, как показано ниже на рис. 1 – энергетической диаграмме реакции (в новом издании [2] это учтено). Здесь А и М – условные обозначения исходных веществ и конечных продуктов, а АК – условное обозначение промежуточного продукта с участием катализатора. Соответственно, есть минимум два переходных состояния, ПС1 и ПС2 (а ПС0 – это переходное состояние без катализатора). В случае системы последовательных реакций энергия активации получения конечного продукта определяется самым высоким из промежуточных потенциальных барьеров. В примере на рисунке это ПС1, но в общем случае это не обязательно барьер первой стадии.
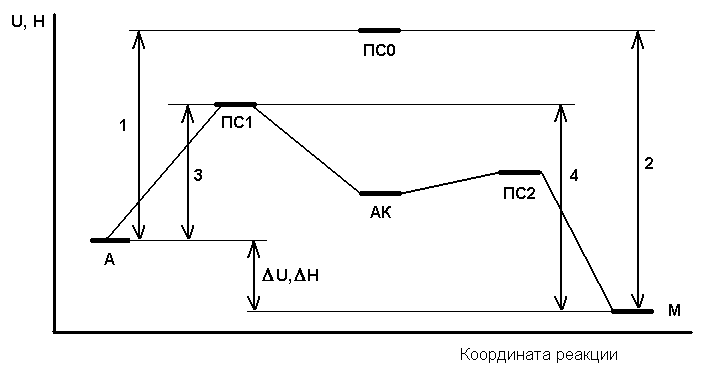
Рис. 1. Энергетическая схема реакции А → М без катализатора
и с катализатором К.
На рис.1 отрезки 1 и 2 – это энергии активации прямой и обратной реакции без катализатора, 3 и 4 – то же с катализатором, ΔU или ΔH – тепловой эффект реакции, выраженный, как изменение внутренней энергии U или энтальпии H (см. § 12.2). В любом случае Еа(прямой) – Еа(обратной) = ΔU или ΔH. На данной схеме реакция экзотермическая (ΔU, ΔH < 0), но это не обязательно.
Принципиально важно, что введение катализатора в любом случае снижает энергию активации и прямой, и обратной реакции на одинаковую величину, и поэтому увеличивает скорость и прямой, и обратной реакции в одинаковое число раз, так что соотношение скоростей в каждый момент остаётся неизменным. В частности, в состоянии равновесия, когда скорость прямой реакции равна скорости обратной, введение катализатора не нарушает этого состояния. Катализатор не смещает равновесие, а лишь ускоряет его достижение.
Во-вторых, важно то, что многие катализаторы действуют избирательно, т.е. ускоряют только одну из нескольких возможных параллельных реакций, причём не всегда самую термодинамически выгодную. Таким образом, правильный подбор катализатора позволяет направлять процессы по желаемому пути.
В третьих, неверно называть ингибиторы – вещества, замедляющие реакцию – отрицательными катализаторами. Если энергия активации процесса с участием ингибитора выше, чем без него, реакция просто не пойдёт по этому пути, её механизм и кинетика останутся прежними. Если же ингибитор препятствует протеканию реакции, значит, он связывается с реагентами или промежуточными продуктами, т.е. расходуется, и это противоречит определению катализатора. Например, ингибиторы цепных реакций окисления масел – это добавки, связывающие свободные радикалы; ингибиторы коррозии металлов – это вещества, связывающиеся с поверхностью металла и затрудняющие доступ агрессивной среды.
Наконец, есть особый случай каталитических реакций – автокатализ, когда катализатором является продукт реакции. Если обычные реакции (каталитические или без катализатора) быстрее всего идут в начальный момент, а потом замедляются, т.к. расходуются реагенты, то автокаталитические реакции идут с самоускорением. В начальный момент, пока катализатор ещё не образовался, реакция идёт медленно. По мере её протекания накапливается катализатор, и реакция ускоряется. Но по мере расходования реагентов реакция неизбежно в конце концов замедлится и прекратится. Следовательно, скорость автокаталитической реакции в функции времени проходит через максимум, а график зависимости концентраций (или количеств) реагентов и продуктов от времени имеет точку перегиба в соответствующий момент (рис. 2).
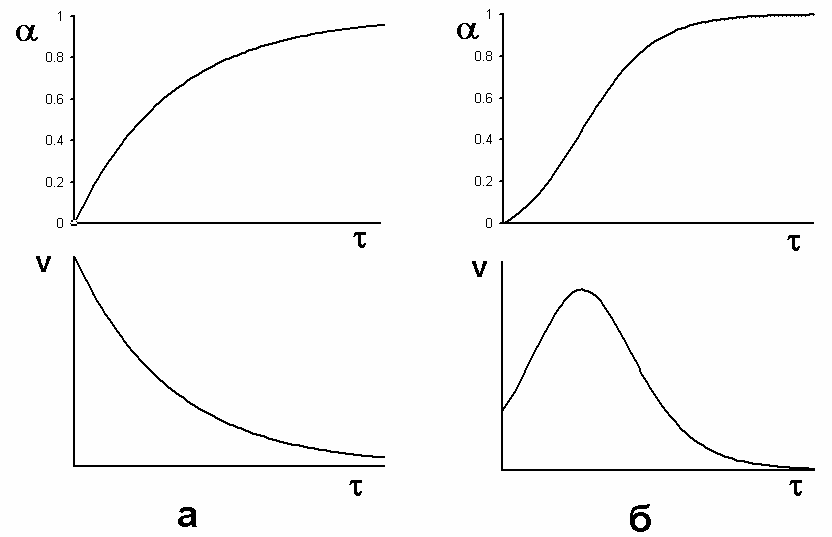
Рис. 2. Так изменяются в функции времени степень превращения α = – ΔC/C0 и скорость реакции v = dα/dτ в случае реакции без катализатора или с заранее введённым катализатором (а) и в случае автокатализа (б). В обоих случаях графики соответствуют реакциям первого порядка.
Примеры автокаталитических реакций:
– растворение меди (или серебра) в растворе азотной кислоты (катализатор – оксид азота): Cu + 4H+ + 2NO3– → Cu2+ + NO2 +2H2O;
– окисление щавелевой кислоты в кислой среде перманганатом калия (катализатор – катион марганца): 5H2C2O4 + 2MnO4– + 6H+ → 10CO2 + 8H2O + 2Mn2+.
Ввиду ограниченного числа лабораторных занятий, практикум по катализу заменяется лекционными демонстрациями. Оба опыта (а) и (б) ниже основаны на разложении пероксида водорода Н2О2 в водном растворе. Это соединение способно практически необратимо разлагаться на воду и кислород: Н2О2 → Н2О + 0,5О2. В самом деле, мы постоянно имеем кислород в контакте с водой, но пероксид из них не образуется. Правда, и Н2О2 может храниться довольно долго из-за высокого потенциального барьера реакции, но на свету, при нагревании, или в присутствии катализатора разложение происходит быстро. Различие в скоростях наглядно без всяких измерений, по выделению пузырьков газа.
(а) Гетерогенный катализ под влиянием крупинки MnO2. То, что это катализатор, а не реагент, доказывается тем, что крупинка в ходе реакции не исчезает, а объём выделившегося кислорода гораздо больше того, который содержится в твёрдом оксиде. Например, если предположить, что MnO2 отдаёт половину содержащегося в нём кислорода, переходя в MnO, то из одного миллиграмма оксида могло бы получиться лишь около 0,1 мл кислорода. А реально выделяются десятки и сотни миллилитров, в зависимости от количества взятого пероксида. Однако в этом опыте не видно признаков того, что катализатор вступает в промежуточные реакции. Поэтому делаем следующий опыт.
(б) Гомогенный катализ под влиянием раствора бихромата калия K2Cr2O7. Этот катализатор не столь активен, поэтому для ускорения процесса используем нагревание в стакане с горячей водой. Но чтобы показать эффект катализа, надо обязательно нагревать и такой же раствор пероксида без катализатора. В данном случае по изменению окраски сразу понятно, что бихромат вступает в реакцию. Чтобы доказать, что он не реагент, а катализатор, нужно обязательно дождаться окончания реакции, когда возвращается исходный цвет бихромата.
В этих опытах может также наблюдаться самоускорение реакции, но не от автокатализа, а от саморазогрева. Мы обсуждаем скорости при одинаковой температуре, а реакция экзотермическая, и если теплота не успевает рассеиваться в окружающую среду, то температура повышается, и это даёт дополнительное ускорение.