

физкал
.pdfСледовательно, если верхняя критическая температура растворения смеси жидкостей лежит выше критической температуры одного из компонентов, то обнаружить ее опытным путем нельзя. Не может быть обнаружена и нижняя критическая температура растворения смеси, если она лежит ниже температуры замерзания одного из компонентов.
В настоящее время существует взгляд, согласно которому кривые расслоения для любой пары жидкостей должны быть замкнутыми. Однако из-за сложности явлений растворимости не создана общая теория взаимной растворимости жидкостей.
IV.4. Взаимно нерастворимые жидкости. Перегонка с водяным паром
Если компоненты бинарной жидкой смеси сильно различаются по своей природе (например, «вода – бензол», «вода – ртуть» и т.п.), то взаимная растворимость их очень мала. Такие жидкости практически не растворяются друг в друге. При постоянной температуре система представлена двумя жидкими слоями из чистых жидкостей и пара над ними. Если жидкости не взаимодействуют между собой, то испарение каждой из них происходит независимо от присутствия другой жидкости, и давление паров каждой из них при постоянной температуре сохраняется постоянным при любых соотношениях масс взятых жидкостей. Поскольку давление пара каждого компонента над смесью практически равно давлению пара над чистой
жидкостью, то по закону Рауля:
робщ = роА + роВ.
где роА и роВ – соответственно, давления насыщенных паров компонентов А и В над чистыми жидкостями А и В при данной температуре.
Эта сумма будет равна внешнему давлению при меньшей температуре, чем температуры кипения индивидуальных жидкостей. Поэтому температура кипения смеси двух таких жидкостей ниже температур кипения каждой из них.
Понижение температуры кипения смеси двух взаимно нерастворимых жидкостей используется при перегонке с водяным паром. К перегонке с водяным паром прибегают в случае жидкостей, практически несмешивающихся с водой и обладающих высокими температурами кипения, а также в тех случаях, когда обычная перегонка невозможна из-за термического разложения перегоняемой жидкости при температурах, меньших ее температуры кипения при атмосферном давлении. Иногда для снижения температуры кипения перегоняемой смеси перегонку производят под вакуумом, т.е. снижая атмосферное давление, но этот способ является более дорогостоящим, так как требует герметичной аппаратуры и применения специальных установок для создания вакуума.
Перегонку с водяным паром проводят следующим образом: в колбу с перегоняемой жидкостью вводят струю водяного пара, в результате чего общее давление пара в системе повышается, и жидкая смесь закипает при более низкой температуре. Пары отгоняемой жидкости вместе с парами воды

поступают в холодильник, в котором конденсируются и стекают в приемник. В приѐмнике эти взаимно нерастворимые жидкости расслаиваются и затем отделяются друг от друга. Соотношение масс жидкостей в конденсате рассчитывают по формуле:
m |
B |
|
р |
M |
B |
|
|
|
|
B |
|
(IV – 4) |
|||
mО |
рО MO |
||||||
|
|
||||||
где рB и рО – соответственно, давления насыщенных паров воды и
перегоняемой жидкости при температуре кипения смеси. Таким образом, состав пара над взаимно нерастворимыми или ограниченно растворимыми жидкостями не зависит от массы жидкостей, а масса перегоняемой жидкости в паре тем больше, чем меньше молярная масса и давление насыщенного пара второй жидкости. Поэтому в качестве второй жидкости при перегонке обычно используется вода, имеющая относительно высокую температуру кипения 100оС и невысокую молярную массу, равную 18 г/моль. Отношение
m B называется расходным коэффициентом, который численно равен массе mO
водяного пара, необходимой для перегонки единицы массы перегоняемого вещества. Величина его тем больше, чем меньше давление насыщенного пара
перегоняемой жидкости ( рО ) и чем меньше еѐ молярная масса (МO).
При помощи водяного пара отгоняют бензол из каменноугольной смолы, очищают анилин и нафталин от примесей; отделяют эфирные масла, извлекаемые из цветов.
IV.5. Трѐхкомпонентные системы. Закон распределения. Экстракция
Если в равновесную систему, состоящую из двух несмешивающихся жидкостей, ввести третье вещество, растворимое в этих жидкостях, то частицы этого вещества распределяются между двумя жидкими фазами в соответствии с законом распределения, установленным в 1890г. В. Нернстом и детально разработанным применительно к различным системам Н. А. Шиловым.
В тех случаях, когда третье вещество 3 находится в одинаковом молекулярном состоянии в обеих равновесных фазах (несмешивающихся жидкостях 1 и 2), термодинамическое равновесие между двумя жидкими фазами наступает при равенстве химических потенциалов третьего вещества в обеих фазах:
|
|
|
|
|
|
|
|
1 |
|
|
2 |
, |
|
|
|
|
|
|
|
|
|
|
|
|
μ μ |
|
|
|
|||||
|
|
|
|
|
|
|
|
3 |
|
3 |
|
|
|
|
||
или при постоянной температуре: |
|
μ1 |
+ RТln а 1 |
= μ 2 + RТln а 2 |
||||||||||||
|
|
|
|
|
|
|
|
|
|
3 |
|
|
|
3 |
3 |
3 |
|
|
|
|
|
μ2 μ1 |
|
|
|
|
|
|
μ 2 |
μ 1 |
|
|
|
|
|
a 1 |
|
|
|
а |
1 |
|
|
3 |
3 |
|
|
|||
|
|
|
|
|
|
е |
RT |
K |
|
|||||||
|
3 |
|
3 |
3 |
|
|
|
3 |
|
|
||||||
откуда |
ln |
|
|
|
|
или |
|
|
|
|
|
|
(IV – 5) |
|||
a 2 |
|
RT |
|
|
а |
2 |
|
|
|
|||||||
|
3 |
|
|
|
|
|
|
|
3 |
|
|
|
|
|
|
|
Полученное |
|
равенство является |
общим |
выражением закона |
||||||||||||

распределения Нернста – Шилова:
Отношение равновесных активностей третьего компонента в двух взаимно нерастворимых жидкостях есть величина постоянная при постоянной температуре, называемая термодинамической константой распределения.
Если растворы третьего вещества в обеих жидкостях идеальны или предельно разбавлены, то активности можно заменить соответствующими концентрациями данного вещества в этих фазах:
С1 |
К |
(IV – 6) |
|
||
С2 |
|
|
Величина К в этом случае называется коэффициентом распределения. Если в двухфазную идеальную равновесную систему ввести несколько
веществ, то каждое из них распределится между двумя нерастворимыми жидкостями в соответствии со своим коэффициентом распределения при данной температуре.
Закон Нернста – Шилова в приведенной выше форме (IV – 6) применим лишь в том случае, если концентрация распределившегося вещества в каждой фазе невелика, и когда это вещество находится в одинаковом молекулярном состоянии и не влияет на взаимную растворимость жидкостей. Если вещество, распределившееся между двумя жидкими фазами, образует ассоциаты или диссоциирует в одной или обеих фазах, то закон распределения имеет другой вид:
К |
С1 |
(1 α) |
(IV – 7) |
|||
|
|
|
|
|
||
n С |
2 |
|
||||
|
|
|
||||
|
|
|
|
|
|
|
где α – степень диссоциации вещества в первой фазе; n – число его молекул, объединяющихся в ассоциированный комплекс во второй фазе. Это уравнение является общим аналитическим выражением закона распределения.
Закон распределения позволяет по опытному значению коэффициента распределения данного вещества определить степень его диссоциации или ассоциации в данном растворителе, вычислить константу равновесия реакции, протекающей в той или иной фазе, а также позволяет определить наиболее выгодные условия извлечения какого-либо вещества из смеси соответствующим растворителем. Процесс извлечения вещества, растворѐнного в одном растворителе, другим растворителем (экстрагентом), который не смешивается с первым и лучше растворяет извлекаемое вещество, называется экстракцией. Пусть в объеме V1 первого растворителя содержится какое-либо вещество массой m, которое надо извлечь при помощи второго растворителя, не смешивающегося с первым. Предположим, что в добавленный объем второго растворителя V2 перейдет (m – m1) вещества, то есть в первом растворителе останется m1 вещества. Если ни в одном из растворителей вещество не диссоциирует и не ассоциирует, то равновесные концентрации его в каждом из растворителей будут соответственно равны:
С |
m1 |
|
|
|
m m1 |
, следовательно, K |
C |
|
|
m V |
||
|
;С |
2 |
|
|
1 |
|
1 2 |
. |
||||
|
|
|
||||||||||
|
|
|
|
|
|
|||||||
1 |
V1 |
|
|
V2 |
|
C |
|
|
V |
(m m ) |
||
|
|
|
|
|
2 |
|
||||||
|
|
|
|
|
|
|
|
1 |
1 |
|
||
Масса вещества, оставшееся не извлеченным из первого растворителя, будет равна:
КV1(m – m1) = m1V2 |
|
KV1m = m1V2 + KV1m1 |
|||
KV1m = m1 (KV1 + V2) |
|
||||
|
|
K V1 |
|
|
|
m1 |
|
|
|
m |
(IV – 8) |
|
|||||
|
|
|
|||
|
K V1 V2 |
|
|
|
|
Данное уравнение позволяет рассчитать количество неэкстрагированного вещества при однократной экстракции. Если после отделения при помощи делительной воронки первого растворителя от второго растворителя повторить операцию с первым растворителем, то в прибавленный объем второго растворителя V2 перейдет m1 – m2 извлекаемого вещества, а в первом останется m2 данного вещества. В этом случае экстракция носит название дробной экстракции. Если данную операцию произвести n раз, то масса неизвлечѐнного из первого растворителя вещества тогда будет равна mn. Представленное выше уравнение (IV – 8) в этом случае будет иметь вид:
|
K V1 |
n |
|
|
|
|
|
|
|
m |
|
(IV – 9), |
|
|
|
|||||
mn |
|
|
|
|||
K V1 V2 |
|
|
|
|
|
|
где n – количество повторений экстракции. Так как |
K V1 |
|
< 1, то чем |
|||
K V V |
||||||
|
|
|
|
1 |
2 |
|
больше n, тем меньше mn, т.е. тем полнее произошло экстрагирование. Отсюда вытекает вывод, что данным объѐмом растворителя можно значительно полнее извлечь вещество из смеси, если производить экстракцию не сразу всем объемом этого растворителя, а последовательно несколькими порциями.
Экстракция широко применяется во многих областях техники и в лабораторных исследованиях. На экстракции основано извлечение сахара в процессе его промышленного производства из свеклы, дубильных веществ и канифоли; очистка и разделение многих нефтепродуктов. При экстракционном методе достигается более полное извлечение масел из семян, чем при механическом прессовании. Экстракция применяется при получении анилина путѐм извлечения его из водных растворов. В последнее время метод экстракции получил широкое применение в аналитической химии. Он позволяет повысить чувствительность определений многих веществ.
V.1. Введение
В конце XIX века наметились два направления в изучении растворов – физическое и химическое. Согласно первому из них растворы рассматривались как чисто физические системы, в которых растворитель играет роль индифферентной среды, а растворенное вещество распределяется по всему объему раствора аналогично газу. При исследовании свойств веществ в очень разбавленных растворах с целью объяснения законов химического равновесия Вант-Гофф нашѐл почти абсолютное тождество физических свойств растворов и идеальных газов. Он показал, что между явлениями растворения и испарения веществ существует глубокая аналогия. Чистая вода или очень разбавленный раствор играют роль пустоты или разреженного пространства для растворенного вещества, которое стремится занять по возможности больший объем. В связи с этим родилась мысль применить законы газообразного состояния, достаточно детально разработанные к этому времени, к разбавленным растворам. Поэтому теория растворов Вант-Гоффа явилась естественным следствием теории Ван-дер- Ваальса о жидком и газообразном состояниях веществ.
Благодаря теории разбавленных растворов Вант-Гоффа оказалось возможным связать в одну стройную систему целый ряд разрозненных наблюдений, произведенных различными методами и являющихся результатом упорной работы нескольких поколений ученых. При этом в отдельную группу были выделены некоторые свойства растворов, которые получили название коллигативных свойств.
Коллигативными называются свойства растворов, которые зависят только от концентрации растворенного вещества, т.е. от числа частиц растворѐнного вещества в единице объема, но не зависят от свойств этих частиц. Этими свойствами являются:
1)относительное понижение давления насыщенного пара растворителя над раствором;
2)повышение температуры кипения раствора по сравнению с чистым растворителем;
3)понижение температуры замерзания раствора по сравнению с чистым растворителем;
4)осмотическое давление раствора.
Коллигативные свойства проявляются наиболее четко в том случае, когда в равновесии находятся две фазы, одна из которых раствор, а вторая – только растворитель.
V.2. Относительное понижение давления насыщенного пара растворителя над раствором
Жидкое состояние вещества характеризуется тем, что молекулы вещества находятся в постоянном хаотическом тепловом движении. В результате этого движения некоторые молекулы отрываются от поверхности

жидкости и переходят в пар, то есть испаряются. Одновременно с этим часть молекул пара, сталкиваясь с поверхностью жидкости, переходят в неѐ, то есть конденсируются.
Вначале скорость испарения жидкости превосходит скорость конденсации, так как число молекул вещества в паре мало. По мере увеличения числа молекул в паре скорость конденсации пара увеличивается и в какой-то момент становится равной скорости испарения жидкости. Между жидкостью и паром устанавливается равновесие, которое характеризуется определѐнным давлением насыщенного пара – рo, которое зависит только от температуры и природы жидкости.
Если растворить в жидкости А какое-либо нелетучее вещество В, то в результате концентрация и поверхность испарения исходной жидкости уменьшится, а значит уменьшится и скорость еѐ испарения, что приведѐт к дополнительной конденсации части молекул пара и установлению нового равновесного давления – р, которое окажется меньше рo. Количественно это явление, как было рассмотрено ранее, описывается законом Рауля:
относительное понижение давления насыщенного пара растворителя над идеальным раствором численно равно молярной доле растворённого вещества в растворе.
р |
|
рА |
робщ |
хВ |
(IV – 2а) |
|
|
р |
|||
р |
|
|
|||
А |
А |
|
|
||
Этот закон точно выполняется для предельно разбавленных растворов неэлектролитов. Но на практике он часто используется и для более широкого интервала их концентраций.
V.3. Повышение температуры кипения раствора неэлектролита
Любая жидкость (чистый растворитель и раствор) закипает при такой температуре, при которой давление насыщенных паров над данной жидкостью станет равным внешнему давлению. На приведенной диаграмме кривая ОА отражает зависимость давления насыщенного пара чистого растворителя от температуры. Абсцисса точки пересечения этой кривой с изобарой в 1 атм. соответствует температуре кипения чистого растворителя
(Тo). Так как давление пара растворителя над раствором при всех температурах ниже давления пара над чистым растворителем, то кривая ВС, отражающая зависимость парциального давления пара растворителя над раствором от температуры, лежит ниже кривой ОА. Если растворѐнное вещество нелетучее, то при Тo давление пара над раствором меньше 1 атм., и поэтому кипения раствора при этой температуре не наблюдается. Чтобы повысить давление

пара растворителя (а в нашем случае и общего давления пара) над раствором, необходимо повышать температуру до какой-то величины Т, которая соответствует достижению кривой ВС значения атмосферного давления. Таким образом, при одном и том же внешнем давлении раствор кипит при более высокой температуре, чем чистый растворитель. Разность между температурами кипения раствора Т и чистого растворителя Тo при постоянном давлении называется повышением температуры кипения раствора:
ТК Т Т .
Эта величина зависит от природы растворителя и концентрации растворенного вещества, но не зависит для разбавленных растворов неэлектролитов от природы растворенного вещества. Следует отметить, что при температуре Т в пар переходят только молекулы растворителя, что приводит к повышению в растворе концентрации нелетучего растворѐнного вещества. Вследствие этого давление пара растворителя (а значит и общего давления пара) над раствором понижается ещѐ больше, и кипение прекращается. Чтобы процесс кипения раствора не прекращался, температуру раствора необходимо постоянно повышать.
Для разбавленных растворов неэлектролитов повышение температуры кипения может быть рассчитано по формуле:
ТК КЭСm |
(V – 1) |
где КЭ - эбулиоскопическая постоянная растворителя или моляльное повышение температуры кипения раствора, так как формально КЭ равна повышению температуры кипения раствора с моляльностью 1 моль/кг. Эбулиоскопическая постоянная может быть рассчитана по формуле:
R (T )2
КЭ Н (V – 2)
исп
где – удельная теплота испарения растворителя, Дж/кг; Тo – температура кипения чистого растворителя. Из выражения (V – 2) следует, что значение эбулиоскопической постоянной определяется только природой растворителя и не зависит от природы растворѐнного вещества. Эбулиоскопические постоянные для различных растворителей приводятся в
справочниках. Для воды КЭ равна 0,516 К кг .
моль
V.4. Понижение температуры замерзания раствора неэлектролита
Любая жидкость (чистый растворитель или раствор) замерзает при такой температуре, при которой давление насыщенного пара растворителя над жидким раствором станет равным давлению пара растворителя над твердой фазой. На приведенной диаграмме кривая ОА отражает зависимость от температуры давления насыщенного пара растворителя над жидким, а кривая OF – над твѐрдым растворителем. В точке О эти давления равны, поэтому абсцисса этой точки (Тo) является температурой кристаллизации

(замерзания) чистого растворителя. Кривая ВС, отражающая зависимость от температуры давления пара растворителя над раствором, в соответствии с законом Рауля, лежит ниже кривой ОА и при Тo не имеет общих точек с кривой OF, поэтому кристаллизации раствора при этой температуре не наблюдается. Пересечение кривых OF и ВС происходит в точке В, абсцисса которой соответствует температуре кристаллизации раствора Т, которая численно меньше Тo. Таким образом, растворы замерзают при температуре более низкой, чем чистые растворители.
Разность между температурами замерзания растворителя и раствора называется понижением температуры замерзания раствора:
∆ТЗ = Тo – Т Необходимо отметить, что при охлаждении разбавленного раствора
вещества, не образующего с растворителем твердых растворов, при температуре, называемой температурой замерзания Т, из него кристаллизуется только твердый растворитель. Поэтому концентрация растворѐнного вещества в растворе увеличивается, давление пара растворителя над раствором понижается ещѐ больше, и процесс кристаллизации при температуре Т прекращается. Поэтому в отличие от чистого растворителя, замерзающего при определенной постоянной температуре, раствор замерзает в некотором интервале температур, то есть для полной кристаллизации раствора температуру системы необходимо постоянно понижать.
Для разбавленных растворов неэлектролитов понижение температуры замерзания раствора может быть рассчитано по формуле:
ТЗ КкрСm (V – 3)
где Ккр - криоскопическая постоянная растворителя. Как и эбулиоскопическая постоянная, Ккр равна понижению температуры замерзания раствора с моляльностью1 моль/кг. Криоскопическая постоянная может быть рассчитана по формуле:
|
R (T )2 |
|
|
Ккр |
|
(V – 4) |
|
Нпл |
|||
|
|
где ∆Нпл – удельная теплота плавления растворителя, Дж/кг; Тo – температура кристаллизации чистого растворителя. Значения Ккр также зависят только от природы растворителя и приведены в справочниках. Для
К кг
воды Ккр = 1,86 моль .
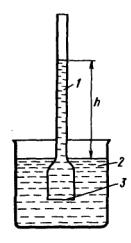
V.5. Осмотическое давление раствора неэлектролита
Осмос – это самопроизвольное проникновение растворителя в более концентрированный раствор, отделенный от него полупроницаемой перегородкой, через которую может проникать растворитель, но не может проходить растворенное вещество.
Для изучения явления осмоса используются приборы – осмометры, схема одного из которых изображена на рисунке. Внутренний сосуд 1 заполнен раствором и отделѐн полупроницаемой перегородкой 3 (полупроницаемой мембраной) от сосуда 2, в котором находится чистый растворитель. В качестве полупроницаемой мембраны может выступать целлофан, вискоза и разные плѐнки из высокомолекулярных веществ. Химический потенциал растворителя в чистом растворителе больше, чем его химический потенциал в растворе. Поэтому в системе начнется процесс, приводящий к выравниванию этих потенциалов. Растворитель будет преимущественно переходить через полупроницаемую перегородку в раствор, что приведет к разбавлению раствора и увеличению его объѐма. Вследствие этого уровень жидкости в трубке сосуда 1 будет повышаться, и будет возрастать гидростатическое давление на полупроницаемую мембрану. Наконец, при некоторой высоте (h) столба раствора скорости диффузии молекул растворителя из сосуда 2 в сосуд 1 и обратно сравняются, и подъѐм жидкости в сосуде 1 прекратится. Гидростатическое давление, которое надо приложить к раствору, чтобы остановить процесс осмоса, называют осмотическим давлением П. Проникновение растворителя в раствор можно предотвратить, если к раствору приложить давление, достаточное для приведения системы в равновесное состояние. Исходя из этого, существует ещѐ одно определение осмотического давления: осмотическое давление – это давление, которое нужно приложить в процессе осмоса к раствору, чтобы привести его уровень к уровню чистого растворителя.
Величина осмотического давления для разбавленных растворов неэлектролитов рассчитывается по уравнению Вант-Гоффа:
П |
n |
RT Cм RT |
(V – 5) |
|
V |
||||
|
|
|
где См – молярная концентрация раствора, моль/м3; П – осмотическое давление раствора, Па.
Из сравнения уравнения Клапейрона-Менделеева и уравнения Вант-Гоффа следует ещѐ одна формулировка осмотического давления, которая гласит, что осмотическое давление разбавленного раствора неэлектролита равно тому давлению, которое оказывало
бы данное количество растворенного вещества на стенки сосуда, если бы оно в виде идеального газа при той же температуре занимало объем, равный объему данного раствора (закон Вант-Гоффа).

V.6. Определение молярных масс веществ криоскопическим и осмометрическим методами
Так как понижение температуры замерзания и повышение температуры кипения растворов неэлектролитов не зависят от природы растворенного вещества, а зависят только от их концентраций, то можно определить молярную массу растворенного вещества, определив экспериментально понижение температуры кристаллизации или повышение температуры кипения растворов с известной концентрацией данного растворѐнного вещества. Метод, основанный на измерении понижения температуры замерзания раствора, называется криоскопическим (криометрическим). Его используют при определении молярных масс растворѐнных веществ, концентрации растворов. Метод, основанный на измерении повышения температуры кипения раствора, называется эбулиоскопическим (эбулиометрическим) методом. Он используется при тех же определениях, что и криоскопический метод. Однако его можно применять только тогда, когда растворенное вещество нелетучее, т.е. его давление пара близко к нулю, и оно не разлагается при температуре кипения. Поэтому для определения многих органических веществ биологического происхождения этот метод не пригоден, и для них применяется криоскопический метод.
Предположим, что gB граммов вещества В с молярной массой МВ растворено в gA граммах растворителя. Число молей В равно gB/MB,
моляльность |
полученного |
раствора |
С |
|
|
1000 gB |
. Тогда |
|
для |
понижения |
|||||||
m |
|
|
|
||||||||||||||
|
|
|
|
|
|
|
gA MB |
|
|
|
|
|
|
||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||
температуры |
замерзания |
раствора |
можно записать: Т |
|
К |
кр |
|
1000 gв |
. |
||||||||
З |
|
||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
gA Mв |
|||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||
Измерив экспериментально ∆ТЗ, можно рассчитать МВ |
растворѐнного |
||||||||||||||||
неэлектролита: |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
M |
|
|
Ккр |
1000 g |
в |
. |
|
|
|
|
|
|
|||
|
|
в |
|
gA ТЗ |
|
|
|
|
|
|
|
||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||
Молярную массу неэлектролита можно также экспериментально определить, измерив осмотическое давление его раствора известной концентрации. Метод, основанный на измерении осмотического давления раствора, называется осмометрическим. Подставляя в уравнение (V – 5)
вместо количества растворѐнного вещества отношение Mm получаем:
П |
m |
M |
m |
||
|
RT . Отсюда |
|
RT . |
||
|
П V |
||||
M V |
|||||
Таким образом, для того чтобы определить молярную массу растворѐнного неэлектролита, необходимо измерить при температуре Т осмотическое давление П (кПа) раствора, содержащего m грамм растворѐнного вещества в V литрах раствора.