

Книги по МРТ КТ на английском языке / MR Imaging in White Matter Diseases of the Brain and Spinal Cord - K Sartor Massimo Filippi Nicola De Stefano Vincent Dou
.pdf178 |
M. Castillo |
may involve all the areas of abnormal white matter including the basal ganglia. In the end-stage disease, brain cysts may form. This cystic leukomalacia may also show contrast enhancement.
12.1.2
Canavan Disease
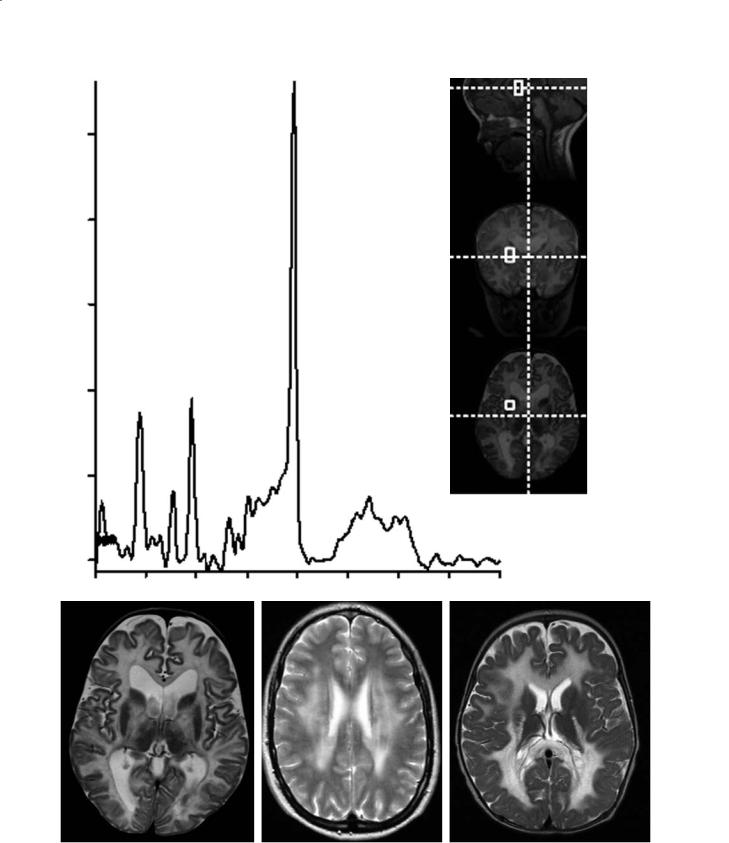
Canavan disease (spongiform leukoencephalopathy) is more common in Ashkenazi Jews,and patients generally die 2–3 years after diagnosis (McAdams et al. 1990).Many patients with macrocephaly and imaging findings identical to those of Canavan disease probably have “van der Knaap disease” (van der Knaap et al.1999).In patients with van der Knaap disease the clinical course is milder and generally only the white matter is diffusely affected. In this disorder, subcortical cysts may be present. Canavan disease is a very rare disorder and is characterized by a deficiency of the enzyme N-acetylaspartylase [which deacetylates N-acetyl aspartate (NAA) to acetate and aspartate], and therefore proton MR spectroscopy shows markedly elevated NAA (Fig. 12.2a). This enzyme is expressed in oligodendrocytes and is involved with the synthesis of myelin lipids. The accumulation of NAA is toxic to the brain and results in myelin splitting (Brismar et al. 1990a). Histologically, there is extensive vacuolization (spongy degeneration) of the brain with increased water content. Canavan disease is autosomal recessive and the gene encoding for acetylaspartylase is located on chromosome 17. Canavan disease generally begins during the first 6 months of life but there are also congenital and juvenile forms of the disease. Most patients die during the first decade of life. There is no effective treatment available. Canavan disease involves all of the white matter in a diffuse fashion including the sub-cortical U-fibers (Fig. 12.2b). In addition, the globi pallidi and thalami are nearly always affected (they show high T2 signal). Much of the affected white and gray matter also show restricted diffusion on diffusion-weighted imaging. No contrast enhancement is present. As the disease progresses, the size of the head begins to decrease.
Another disease that needs to be included in the differential diagnosis of Canavan disease is Cree leukoencephalopathy. The imaging findings are identical to those of Canavan but Cree has been reported only in northern Quebec and Manitoba in Canada (Alorayni et al. 1999). Patients with congenital muscular dystrophy of the merosin-deficient type may also present with diffuse increased T2 signal intensity in the brain (Fig. 12.2c). These patients, however,
have near normal psychomotor development (compared to those with Canavan disease), have muscle weakness of early onset and may have seizures. In both of the previously mentioned disorders, the head size is not enlarged. Diffuse white matter involvement may also be seen in the rare “vanishing brain syndrome” (Fig. 12.2d).
12.1.3
Krabbe Disease
This is also a very rare disorder, although in the author’s experience it is perhaps somewhat more common than Alexander and Canavan diseases. It is also called globoid cell leukodystrophy. It is a lysosomal disorder characterized by a deficiency of the enzyme galactocerebroside beta galactosidase, and is inherited as an autosomal recessive trait (Hittmair et al. 1994). The enzymatic defect blocks the degradation of beta galactocerebroside, which is a critical component of the myelin sheaths. The presence of end products such as psychosine is toxic to the oligodendrocytes. The diagnosis is confirmed by establishing the levels of leukocyte lysosomal beta galactosidase (Choi and Enzmann 1993). Krabbe disease generally affects infants and young children, and most patients are diagnosed between the 3rd and 6th month of life (one of the earliest leukodystrophies). The head is initially large and then becomes small (at presentation most patients are microcephalic). CT may show increased density in the thalami but, since this study is nowadays seldom obtained for patients suspected of having leukodystrophies, this finding is not commonly observed (Finelli et al. 1994). MRI shows increased signal intensity around the ventricles in a non-spe- cific pattern (Hittmair et al. 1994). High T2 signal may also be seen in the pyramidal tracts and in the medial aspect of the cerebellar hemispheres. In the late onset variant, the splenium of the corpus callosum may be involved. Atrophy ensues in most patients, particularly late in the disease. The brain abnormalities generally do not show contrast enhancement. Krabbe disease may result in hypertrophy of the cranial and spinal nerves (simulating the findings seen in Guillain-Barrè syndrome) (Lee et al. 1995). These large nerves may show contrast enhancement. Bone marrow transplantation may be helpful in the treatment of the disease. Diffusion tensor imaging may be the best way to evaluate these patients. Tensor imaging shows more abnormalities than T2 weighted imaging and provides a quanti-
Imaging of Inherited and Acquired Metabolic Brain Disorders |
179 |
250
200
150
100
50
0
a |
4.0 |
3.5 |
3.0 |
2.5 |
2.0 |
1.5 |
1.0 |
0.5 |
0.0 |
b |
c |
d |
Fig. 12.2a–d. Canavan disease and mimics. a Long echo time MR spectroscopy obtained from the white matter shows that the peak of N-acetyl aspartate (2.0 ppm) is markedly elevated. b Axial T2-weighted image in the same child shows diffuse abnormal hyperintensities throughout the white matter and also in the globi pallidi and thalami. c In a child with merosin-deficient muscular dystrophy, axial T2-weighted image shows abnormal signal intensity from the white matter at the level of corona radiata. d In this child with the vanishing brain syndrome, there is abnormal T2 hyperintensity in all of the white matter

180 |
M. Castillo |
tative measurement of the severity of the disease, making it an optimal way to study the response to therapy.
12.1.4 Mucopolysaccharidoses
This group of disorders also affects the white and the gray matters, and is included here because children may also present with an enlarged head (in reality the diagnosis is well established in most patients by the time of imaging evaluation). They are not true leukodystrophies,but glycosaminoglycans are deposited along the perivascular spaces, leading to focal changes mostly in the periventricular white matter. These compounds are also accumulated inside the cells and are toxic to them. The mucopolysaccharidoses may be considered as disorders of lysosomes (Johnson et al. 1984). Initially there is macrocephaly but progression of these disorders eventually results in brain atrophy. All are inherited in an autosomal recessive fashion, except for Hunter’s disease which is x-linked. The most common mucopolysaccharidoses are Hurler, Morquio, Hunter, while the least common are Sanfilippo and Maroteaux-Lamy disease. The life span of these patients is variable,but is now improved with the availability of bone marrow transplantation and replacement with recombinant human enzyme (L-iduronidase). The skull is thick and there is doli-
chocephaly and a thick and somewhat keel-shaped frontal bone (these are prominent features in Hurler disease). Expansion of the skull may lead to multiple cranial nerve deficits. Ventricular dilatation with or without increased intracranial pressure is common (Kumar et al. 1987). It is often very difficult to tell whether there is increased intracranial pressure due to the ventricular dilatation. The white matter may show abnormal T2 signal intensity and may contain multiple dilated peri-vascular spaces (Fig. 12.3). It is not clear if these peri-vascular spaces are dilated by the glycosaminoglycans or are part of the spectrum of the communicating hydrocephalus. In the author’s experience, their signal intensity is identical to that of cerebrospinal fluid in all MR imaging sequences, including FLAIR and DWI. The presence of multiple dilated peri-vascular spaces involving the corpus callosum is typical of the mucopolysaccharidoses (in tuberous sclerosis there are multiple dilated perivascular spaces, but they almost never involve the corpus callosum and, when they do, they are few and isolated). The cortical sulci may be prominent due to brain atrophy, and occasionally one or more arachnoid cysts are presumably present secondary to the deposition of glycosaminoglycans in the meninges. Little correlation, however, has been seen by the author between the patient’s clinical deficits and the imaging findings, that is, the brain may be severely involved but the patient may have only mild clinical manifestations of the disease.
a |
b |
c |
Fig. 12.3a–c. Hunter disease. a Axial T2-weighted image shows dilated perivascular spaces in the posterior periventricular white matter. b Corresponding FLAIR image shows that the lesions are isointense to the cerebrospinal fluid. c There are also dilated perivascular spaces in the peri-trigonal white matter and in the thalami

Imaging of Inherited and Acquired Metabolic Brain Disorders |
181 |
12.2
Predominantly White Matter Disorders with Normal Head Size
12.2.1 Adrenoleukodystrophy
Adrenoleukodystrophy (ALD) is the most important peroxisomal disorder and one of the most common primary white matter disorders in children. The most common type of these disorders is inherited as an x- linked recessive trait (Xq28) and thus it is only seen in males (Jensen et al. 1990). The neonatal form of ALD is inherited as an autosomal recessive trait and is very rare. Other types of the disease include the adrenomyeloneuropathy that affects older individuals and the presymptomatic ALD, in which the genetic defect is present but imaging and clinical abnormalities are nearly absent. The affected gene in x-linked ALD encodes for the formation of coenzymes derived from the fatty acids within the peroxisomes. This leads to impaired oxidation of fatty acids and their accumulation, which is detrimental to the formation and stabilization of myelin, resulting in active demyelination and inflammation. Very long chain fatty acids are not correctly metabolized and become elevated in serum and other tissues. The defect is at the level of the lignoceroyl coenzyme A ligase. X-linked ALD occurs in boys of 3–10 years of age and a vegetative state or death may occur as soon as 2 years after the diagnosis (Snyder et al. 1991). The most common clinical symptoms are sensorineural hearing loss (90%),pyramidal tract symptoms (50%), visual deficits (40%), behavioral abnormalities (33%) and seizures (6%).All patients will become vegetative within 5 years of diagnosis. The only hope for cure is early bone marrow transplantation.
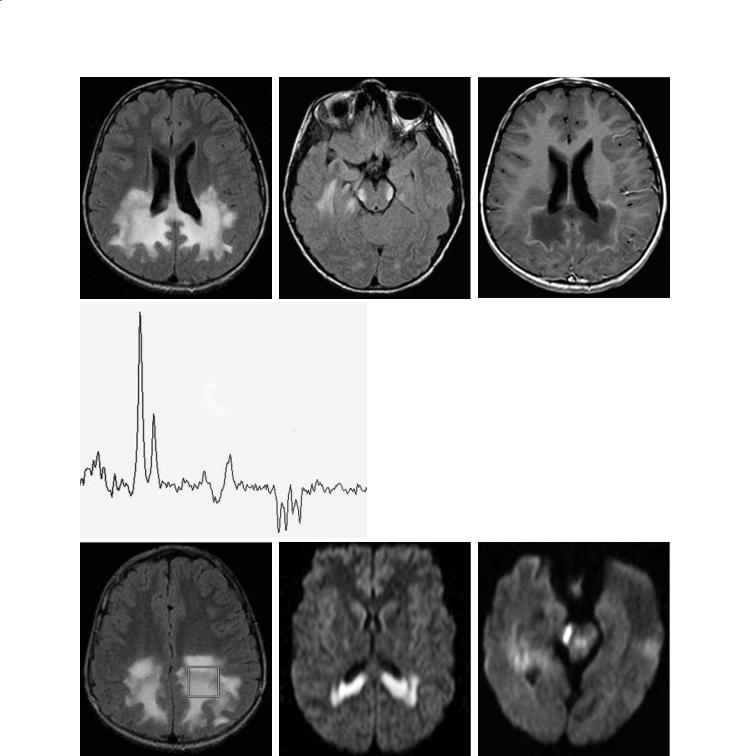
In 80% of cases,CT and MRI show abnormalities in the white matter of the occipital regions, also involving the splenium of the corpus callosum and extending anteriorly (Fig. 12.4a). The location of the abnormalities closely correlates with the symptoms (Loes et al. 1994). Early in the disorder, only the corticospinal tracts and/or the medial lemnisci may be affected (Melhem et al. 1996) (Fig. 12.4b). The anterior border of these abnormalities may enhance after contrast administration and this finding implies breakdown of the blood–brain barrier due to active demyelination and inflammation (Fig. 12.4c). Pathologically, the following changes are seen in ALD: zone A (inner zone) corresponds to scar and gliosis and shows no contrast enhancement and high T2 signal; zone B
(middle zone) corresponds to inflammation and demyelination with axonal preservation and shows contrast enhancement and high T2 signal intensity; and zone C (outer zone) which has active destruction of myelin but no inflammation. This latter zone is very thin and is not clearly visualized.
Proton MR spectroscopy obtained at the level of the disease front (zones B and C) shows significant elevation of the choline secondary to the presence of inflammatory cells. On MR spectroscopy, NAA (a neuronal marker) is decreased, and lactate and lipids (from cellular breakdown) may be elevated (Rajnanayagam et al. 1996, 1997) (Fig. 12.4d). Peaks between 0.9–2.4 may be seen and are believed to be related to the presence of the very long chain fatty acids. Patients with an inflammatory component have a more rapid and severe course and a worse prognosis. Diffusion-weighted images may show restriction of water diffusion early in the course of the disease (Fig. 12.4e,f).ALD rarely begins in the frontal regions. In the chronic stage of the disease, dystrophic calcifications may be seen in the white matter (Barkovich et al. 1994). This finding is not only unusual, but it is seldom seen, because CT is no longer commonly obtained in ALD patients.
12.2.2
Metachromatic Leukodystrophy
Metachromatic leukodystrophy (MLD) is a lysosomal disorder that is characterized by a deficiency in arylsulfatase A (or one of its cofactors) and accumulation of sulfatides that are toxic to the white matter (Kim et al. 1997). There is lack of breakdown and reutilization of myelin and the sulfatides impair the function of macrophages and Schwann cells. The presence of sulfatides in urine and deficient arylsulfatase A in leukocytes confirms the diagnosis. It is the most common leukodystrophy and is inherited as an autosomal recessive trait. There is gene mutation in chromosome 22 but this is not present in all patients. Most patients become symptomatic between 1–3 years of age (late infantile form). There is a less common juvenile form in which the patients generally present between 5–7 years of age. Death occurs 1–4 years after diagnosis. Bone marrow transplantation has been tried with varying degrees of success. On imaging studies, the white matter is diffusely affected but the subcortical u-fibers are spared (Kim et al. 1997). The pattern of involvement is periventricular, bilateral and symmetrical (‘butterfly’ pattern) (Fig. 12.5a). The areas of high T2 signal inten-
182 |
M. Castillo |
a |
b |
c |
d
e |
f |
g |
Fig. 12.4a–g. X-linked adrenoleukodystrophy. a Axial FLAIR image shows increased abnormal signal intensity in the occipitoparietal white matter and crossing the callosal splenium. b In the same patient, a FLAIR image shows symmetrical hyperintensities in the region of the lateral lemnisci. c Post contrast T1-weighted image at same level as (a) shows enhancement of the periphery of the lesions. d MR spectroscopy study obtained with an echo time of 135 ms shows very low NAA, high choline and presence of lactate. e Axial FLAIR image shows location of the voxel used for the MR spectroscopy study shown in (d). f In a different patient, axial trace diffusion-weighted image shows increased signal intensity in the peri-trigonal white matter and in the splenium of the corpus callosum. g In the same patient, there is restricted water diffusion in the region of the right lateral lemniscus
Imaging of Inherited and Acquired Metabolic Brain Disorders |
183 |
a
b 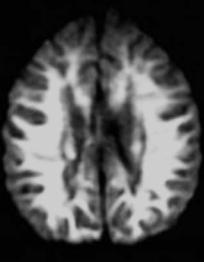
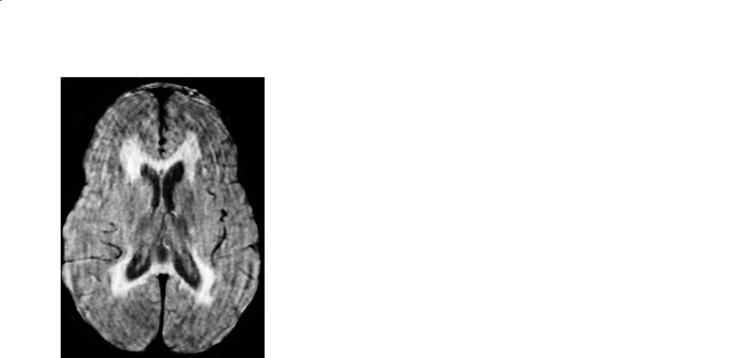
Fig. 12.5a,b. Metachromatic leukodystrophy. a Axial FLAIR image shows non-specific symmetrical periventricular abnormal high signal intensity. b In a different patient, axial trace diffusion-weighted image shows abnormal high signal intensity throughout the white matter
sity may also be somewhat patchy in appearance. There is no contrast enhancement and the areas of abnormal T2 signal also show abnormal diffusion (Fig. 12.5b). The medial regions of the cerebellum may be involved early in the course of the disease. Eventually all of the brain (including corpus callosum and brainstem) become involved and atrophy ensues (Stillman et al. 1994).
12.3
Predominantly White Matter Disorders with Small Head Size
12.3.1
Pelizaeus-Merzbacher Disease
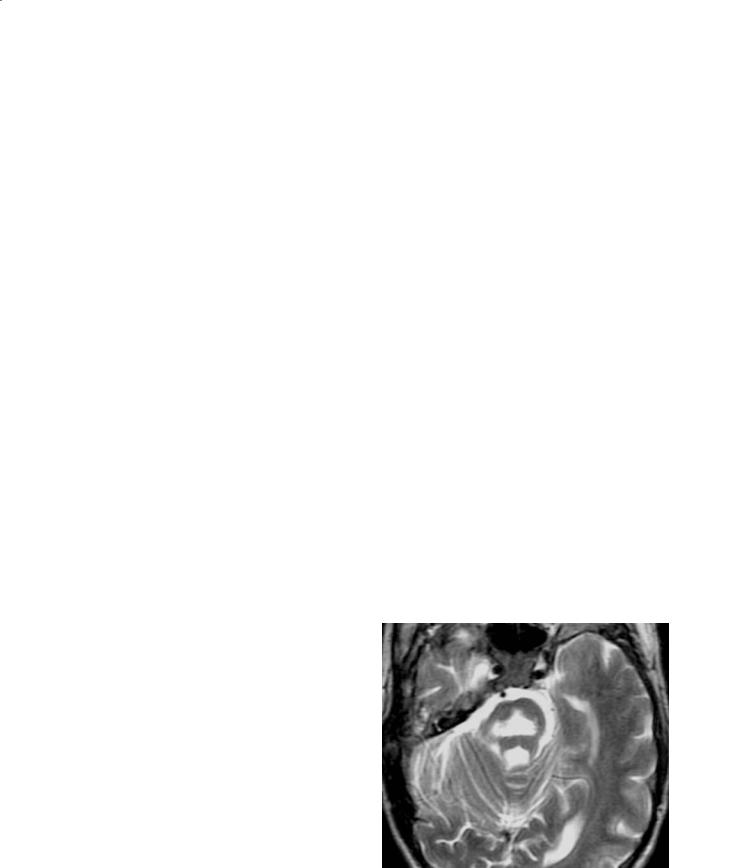
Pelizaeus-Merzbacher disease (PMD) is an x-linked disorder (classic form) that is characterized by the lack of myelin specific lipids leading to abnormal function of the oligodendrocytes and hypomyelination. It belongs to the group of disorders called ‘sudanophilic’ leukodystrophies (Silverstein et al. 1990). PMD presents very early in life. All of the patients seen by the author presented with abnormal eye movements and cerebral palsy-like symptoms. The patients also show developmental delay, seizures, spasticity and all die a few years after the initial diagnosis (Silverstein et al. 1990). The white matter never forms normally due to a defect of the gene that encodes for a ‘proteolipid’ protein, which is essential for the development of myelin.On histology,the white matter has a “tigroid” appearance (van der Knaap and Valk 1989). On imaging studies, the white matter is diffusely abnormal and the brain retains an in utero appearance (the abnormalities may be so diffuse and symmetrical that at first glance the studies may even be interpreted as normal) (Fig. 12.6a,b). There is lack of mature myelin throughout most of the white matter. In the later stages of the disease, the basal ganglia may be dark due to increased iron deposition. The cerebellum may be small and eventually the brain shows atrophy.
Trichothiodystrophy deserves to be briefly mentioned here because the brain may show abnormalities similar to those seen in PMD on MRI. These patients have ectodermal defects such as brittle hair and abnormal dental enamel (Wetzburger et al. 1998). In addition to the usual neurological findings seen in most white matter disorders, patients with trichothiodystrophy have a peripheral neuropathy and abnormal hearing. This disease is extremely rare.
12.4
Osmotic Myelinolysis
Osmotic myelinolysis is an acute demyelinating disorder most often seen 2–3 days after the rapid correction of hyponatremia (<15 mmol/L), but may also occur in hypernatremic states or even in the
184
a 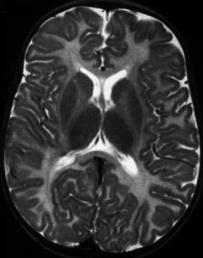
b 
Fig. 12.6a,b. Pelizaeus-Merzbacher disease. a Axial T2weighted image at 3 years of age shows diffuse lack of myelination. b In a different patient, a T1-weighted image shows diffusely low signal intensity from the white matter indicating lack of mature myelin
M. Castillo
altered temperature regulation, seizures, respiratory arrest and coma).
Most patients with osmotic myelinolysis are chronic alcoholics, have extensive burns, sepsis, Hodgkin disease, or other malignancies. Osmotic myelinolysis has a high mortality rate, but currently many patients survive. On imaging, most patients show abnormalities in the pontomedullary junction (called ‘central pontine myelinolysis’ or CPM for short) (Sztencel et al. 1983). Histologically, CPM shows acute demyelination (particularly of the pontine transverse fibers), with preservation of the neurons and axons. Patients with CPM present with lethargy, cranial nerve abnormalities, problems in swallowing, and may progress to quadriparesis, including a ‘lock in’ syndrome.
Imaging studies are reported as normal in up to 85% of patients with the clinical syndrome of CPM. MRI shows an area of high T2 signal centered in the lower pons (Korogie et al. 1993; Price et al. 1987) (Fig. 12.7). Sometimes the lesion has a triangular appearance on axial images with its apex located in the midline and pointing forward (pontine infarcts may be triangular-shaped, but the lesion is nearly always off midline and its apex points posteriorly). In most patients, CPM has a nonspecific appearance and involves the entire cross section of the brainstem. The lesion is devoid of mass effect and contrast enhancement. Although the cerebellar cortex is affected commonly, this finding is not usually appreciated on MRI. In 10% of patients, the supratentorial gray matter structures that are surrounded
absence of sodium concentration abnormalities (Ho et al. 1993). It is possible that rapid influx of water into the cells associated with sodium alterations results in their damage. The neurological symptoms of hyponatremia may be divided as follows: early stage (anorexia,nausea,vomiting,muscle spasm and weakness), advanced stage (impaired responses to verbal commands and pain,bizarre behavior,hallucinations, obtundation, respiratory distress) and severe stage (decortication, bradycardia, hyperor hypotension,
Fig. 12.7. Central pontine myelinolysis. Axial T2-weighted image shows a triangular-shaped area of abnormal high signal intensity in the mid pons

Imaging of Inherited and Acquired Metabolic Brain Disorders |
185 |
by white matter may be affected (extrapontine myelinolysis). The regions most commonly affected are the lentiform nuclei and the thalami. Occasionally, supratentorial white matter structures such as the internal capsule and corpus callosum may be involved. When the patients survive, a slow and gradual decrease in the size of the lesions is noted on follow-up studies, but their lesion does not resolve completely (Rosenbloom et al. 1984).
12.5
Predominantly Gray Matter Disorders
12.5.1
E ects of Liver Failure and Parenteral Nutrition
It is well known that hepatic insufficiency will lead to abnormalities on brain MR imaging. T1 weighted images show increased signal intensity in the caudate nucleus, tectum (particularly the inferior colliculi), globuspallidus,putamen,subthalamicnucleus,rednucleus, adenohypophysis, and substantia nigra (Etsuo et al.1991).The abnormalities are bilateral,symmetrical, and of homogenous signal intensity; there are no corresponding abnormalities on T2 weighted images and CT scans are normal. The MR imaging findings are believed to be due to increased amounts of manganese (Mn) and other factors leading to shortening of the relaxation time. Increased plasma levels of Mn may be found in chronic liver failure, patients receiving long-term parenteral nutrition, and occupational toxicity (Krieger et al. 1995). Nearly 95% of Mn is excreted in bile. Manganese is involved in some enzymatic cell cycles, including superoxide dismutase and glutamine synthetase. Manganese reaches the brain by way of erythrocytes and plasma. Transferrin and albumin are its carriers in plasma. The half-life of blood Mn is 10–42 days, but when it reaches the brain it may remain there for prolonged periods of time. Astrocytes have specific transport systems for Mn. Manganese is neurotoxic and results in striatal dopamine depletion, NMDA excitotoxicity, and oxidative stress (Krieger et al. 1995). Thus, Mn may play a role in the development of hepatic encephalopathy that is clinically characterized by pyramidal and extrapyramidal dysfunction, brisk tendon reflexes, and tremors. The concentration of Mn in the pallidus of cirrhotic patients is three-fold higher compared to controls (Pomier-Layrargues et al. 1995). The frequency of MR imaging abnormalities in the brain
of cirrhotic patients is approximately 73% (PomierLayrargues et al. 1995).
Long-term total parenteral nutrition results in increased T1 signal intensity in the same regions. These solutions are rich in Mn and lead to development of neurological symptoms. Once the parenteral nutrition is discontinued, the MR abnormalities may reverse to normal (Fell et al. 1996). The amount of time required for these abnormalities to resolve is not clear, but most do within 1 year after cessation of parenteral nutrition (Mirowitz and Westrich 1992). T1 hyperintensity in the adenohypophysis and dorsal brainstem due to total parenteral nutrition may revert to normal 4 months after treatment is discontinued (Okamoto et al. 1998). Similar findings are noted in patients following liver transplantation (Krieger et al. 1995). Although the clinical and imaging abnormalities are reversible in adults, its longterm effect on the child’s brain is not known.
Other MR techniques may be more sensitive to the effects of liver disease on the brain than just imaging. Magnetization transfer contrast ratios are abnormal not only in the previously named brain regions but also in the white matter (Iwasa et al. 1999). With liver failure there is an increased proliferation of Alzheimer type II astrocytes in the affected brain regions. The etiology for the proliferation of these cells may be hyperammonemia. These cells are characterized by cytoplasmic hypertrophy and increased water content. This relatively large amount of water may decrease magnetization transfer by virtue of protein dilution. Repeated episodes of hepatic failure may lead to the development of acquired (non-Wilsonian) hepatocerebral degeneration. The clinical symptoms are permanent and death commonly follows. By imaging, these patients no longer show the typical T1 hyperintensities in the basal ganglia described above for chronic hepatic encephalopathy. Patients with acquired hepatocerebral degeneration show increased T2 signal intensity, particularly in the middle cerebral peduncles (Lee et al. 1998). These zones correspond to spongiform myelinolytic changes, most likely the sequelae of ischemia. These changes are not secondary to osmotic myelinolysis.
12.5.2
Bilirubin Encephalopathy
Kernicterus is now a rare disorder and most cases are the result of isoimmunization to Rh factor or from ABO blood group incompatibility. As such, it is almost always a disease of neonates (Rorke 1998).
186 |
M. Castillo |
Later in life, most cases are due to genetic, hematologic, or liver diseases, such as defects of glucose- 6-phosphate-dehydrogenase, Crigler-Najjar disease, Gilbert disease and in rare cases secondary to breast feeding or severe sepsis. In kernicterus, the liver is unable to conjugate bilirubin. The enzyme uridinediphosphate glucuronyl transferase does not function well and albumin-bound (insoluble) bilirubin cannot be deconjugated into bilirubin diglucuronide or“water soluble”bilirubin.Initially the child appears hypotonic, then he/she becomes hypertonic, and after approximately 1 week the hypotonia returns. On gross brain examination, there is yellow staining of the globus pallidus, mammillary bodies, subthalamic nuclei, and the indusium griseum. In the brainstem, the substantia nigra and some of the cranial nerve nuclei may be involved (Rorke 1998).
The histological findings are identical to those caused by hypoxia and/or hypoglycemia and reflect tissue necrosis. Surrounding inflammatory changes are common. MR imaging has been used to evaluate mostly the sequelae of kernicterus (Erbetta et al. 1998). During the chronic stage there is increased T2 signal intensity in the globus pallidus and in the cerebellar dentate nucleus. These structures are also atrophic (Fig. 12.8). Acutely, babies with kernicterus may show increased T1 signal intensity in the basal ganglia without corresponding T2 weighted abnormalities. I believe that this finding is probably related to acute hepatic insufficiency and may be similar to that seen in adults with chronic liver disease.
Fig. 12.8. Effects of liver failure. Axial T1-weighted image shows increased symmetrical signal intensity in the lentiform nuclei
12.5.3
Wilson Disease
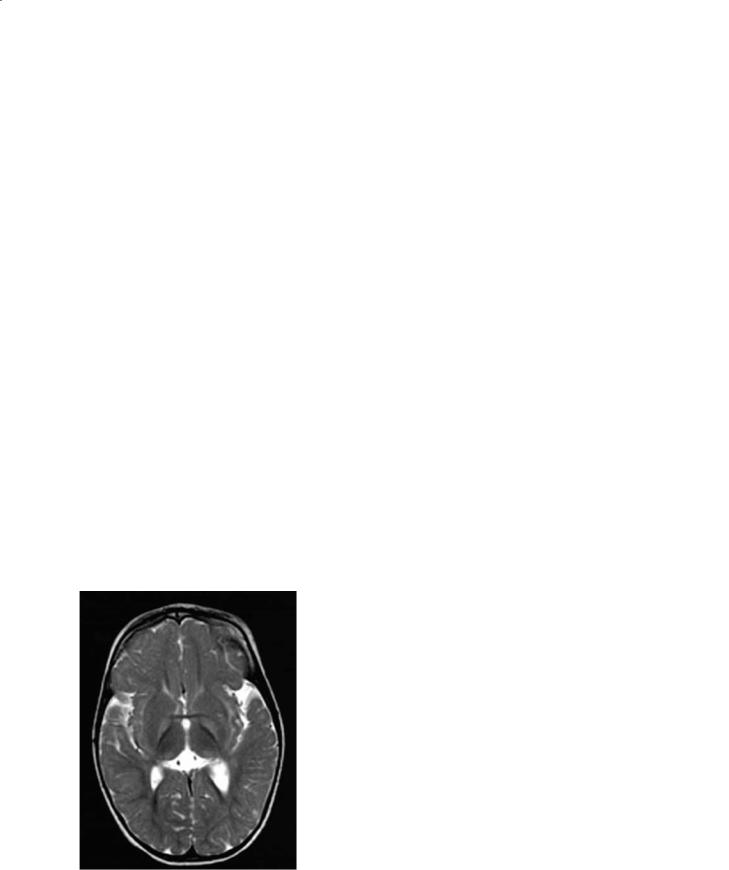
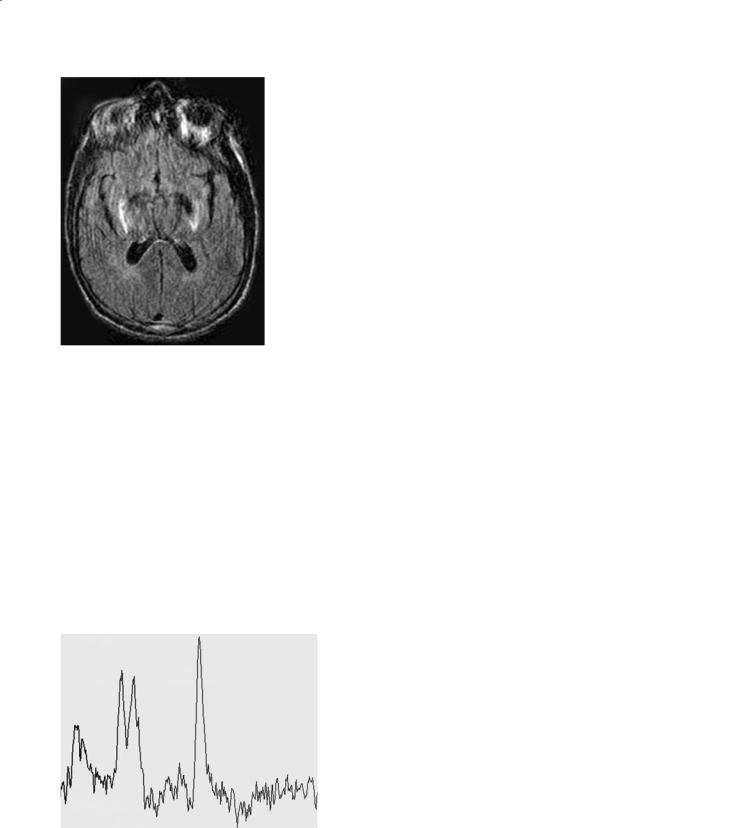
Wilson disease is a genetic disorder of copper metabolism that is characterized by failure to incorporate copper into ceruloplasmin in the liver, failure to excrete copper by the liver into the bile, and toxic accumulation of copper in multiple organs (Albernaz et al. 1997). The defect has been mapped on chromosome 13 q14.3. In addition, concentrations of other heavy metals such as zinc, iron, silver, and aluminum are also increased. The genetic defect is localized to 13q 14.3 and several mutations have been described (Albernaz et al. 1997). Most children present with hepatic failure while most adults present with neurological symptoms. Accumulation of copper within Descemet’s membrane results in the Kayser-Fleischer rings. Accumulation of copper in the lens results in the rare “sunflower” cataracts. Pathologically there is cell loss and cavitation in the lentiform nucleus. Copper concentration at these sites is markedly increased. Other regions involved include the thalamus, subthalamus, red nucleus, substantia nigra, dentate nucleus, and the brainstem. Microscopically, these regions contain large cells with small nuclei (termed Opalski cells) and these are thought to be typical for Wilson disease. Similar to effects of other liver disorders on the brain Alzheimer type II cells are also found in the affect areas. The brain is diffusely atrophic.
MR imaging shows abnormalities even in absence of clinical neurological findings. The paramagnetic effects of copper are visible by MR imaging only in untreated patients (Magalhaes et al. 1994). Basal ganglia lesions are most often bilateral and symmetrical. The putamina shows striking increase in T2 signal intensity (Fig. 12.9a). This is present to a lesser degree in other deep gray matter structures. Thalamic lesions are often present but typically spare the dorsomedial nuclei. White matter tracts including the dentatothalamic, corticospinal, and pontocerebellar tracts are commonly involved. The claustrum may show high T2 signal intensity. The midbrain is bright on T2 weighted images with relative sparing of its deep nuclei giving rise to the so-called Panda sign (Albernaz et al. 1997) (Fig. 12.9b). Although it seems attractive to postulate that high copper concentrations are responsible for the MR imaging findings, this is probably not true. Copper results in T1 and T2 shortening, thus the imaging findings in Wilson disease are probably secondary to spongy degeneration, cavitation, neuronal loss, and reactive astrocytosis. There is a correlation between the clinical and MR
Imaging of Inherited and Acquired Metabolic Brain Disorders |
187 |
a
b 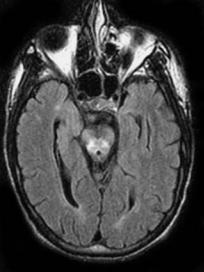
c
Fig. 12.9a–c. Wilson disease. a Axial FLAIR image shows low signal intensity from the globi pallidi and abnormal high signal intensity from the lateral putamina. b In the same patient, there is abnormal high signal intensity in the peri-aqueductal region in the midbrain. c Long echo time MR spectroscopy shows mildly decreased N-acetyl aspartate
imaging findings. An abnormal appearing striatum correlates with pseudoparkinsonism, an abnormal dentatothalamic tract with cerebellar signs, and an abnormal pontocerebellar tract with pseudoparkinsonism. The presence of portosystemic shunting may correlate with abnormalities seen in the globus pallidus. The abnormal T2 signal intensity may improve after copper trapping therapy (King et al. 1996). Contrast enhancement is rare but may be seen when patients are receiving treatment with penicillamine. Positron emission tomography shows diffusely reduced glucose metabolism particularly marked in the caudate nucleus, frontoparietal cortex, and lentiform nucleus (van Wassenaer-van Hall et al. 1996; Engelbrecht et al. 1995). These findings correlate with the severity of the disease and may be reversed following adequate therapy (Hawkins et al. 1987).
12.5.4
Disorders of Iron Metabolism That May Involve the Brain
Although the disorders involving iron metabolism are relatively common (particularly those related to iron deficiencies), their effects on the CNS are few. It is well known that anemia related to chronic and debilitating disorders such as AIDS may result in a spine that appears hypointense on T1 weighted MR imaging. This is presumably due to a compensatory mechanism that leads to storage of iron in the bone marrow. The presence of iron results in a diffuse hypointense appearance of the bone marrow (Kuwert et al. 1992).
Disorders of iron overload are less common than anemias, but occasionally give rise to interesting, albeit uncommon, imaging findings. Iron overload occurs when the iron-binding capacity of transferrin is exceeded or when an increased catabolism of erythrocytes leads to accumulation of iron in the reticuloendothelial and organ cells. Patients with hereditary hemochromatosis have an increased iron intestinal absorption. In the brain, iron is deposited in the pituitary gland and may give rise to abnormal and low endocrine functions. In these patients adenohypophysis may appear as an area of very low or no signal in both T1 and T2 weighted images (Cordato et al. 1998). In a recent article, a ratio of the signal intensity of the pituitary gland to fat was correlated with gland function (Geremia et al. 1989). These authors found that reductions in this ratio correlated with hypogonadotropic hypogonadism and higher ferritin levels. In addition, in these patients the