

Книги по МРТ КТ на английском языке / MR Imaging in White Matter Diseases of the Brain and Spinal Cord - K Sartor Massimo Filippi Nicola De Stefano Vincent Dou
.pdf242 |
J. H. Simon and B.-K. Kleinschmidt-DeMasters |
these terms to denote certain stereotypic sites of involvement irrespective of co-existent disorders. An example of this would be the use of “Devic’s disease” for spinal cord and optic nerve lesions in autoimmune disorders or cancer.
Today, based on neuropathology and imaging, Marburg (acute MS), Schilder (large coalescent, usually childhood, MS with exclusive cerebral hemispheric involvement), and Balo (alternating concentric rings of myelin loss and preservation in MS) are accepted as variants of MS. Devic’s disease (as defined by exclusive spinal cord and optic nerve involvement) on the other hand, from both an imaging and neuropathology perspective is less confidently categorized today as an MS variant, but if it is, it likely lies on the far-side of the spectrum of MS (Table 16.1).
All these variants result in diagnostic difficulty and errors disproportionate to their incidence. A few additional points are worth noting:
1)The initial clinical presentation of these MS variants is usually severe and rapidly progressive, making the differential diagnostic list broad.
2)Making the diagnosis at the very initial phases of any of these diseases (when the neuroradiologist is most likely to be confronted with the disorder by imaging studies) is virtually impossible. Only passage of time and evolution of clinical features of the disease, biopsy, or even autopsy provide the final and correct diagnosis.
3)All these MS variants are rare and heterogeneous within each category due to differing uses of the terms, as noted above. Although correct diagnosis is always desirable, in actuality, treatment decisions are more often guided by anecdote and desperation than by evidence-based studies.
4)Despite these difficulties, from a research perspective, these MS variants as “outliers” do provide important material with which to address questions related to variant host response, mechanisms of remyelination or barriers to remyelination, and issues surrounding new hypotheses of MS classification (Lucchinetti et al. 2000).
This chapter will attempt to outline the unique versus the overlapping clinical, pathological, and neuroimaging features of Marburg, Schilder, Balo, and Devic types of MS. Acute tumefactive demyelinating lesions will also be briefly discussed under the acute MS section.
16.2
Devic’s Neuromyelitis Optica
16.2.1
General Features
Devic’s neuromyelitis optica (DNO), originally described in 1894 (Devic 1894) is an inflammatory demyelinatingdiseasewithfeaturesthatoverlapwithMS (Cree et al. 2002; de Seze et al. 2003; Weinshenker 2003). However, in contrast to the other MS variants discussed below, DNO has come to be understood as more distinct than similar to MS, based on clinical, pathological, immunological and imaging criteria.
The key clinical features of DNO include acute visual loss,often bilateral,and acute transverse myelitis, with the visual and spinal cord signs and symptoms often presenting nearly simultaneously (O’Riordan et al. 1996; de Seze et al. 2003; Wingerchuk et al. 1999). Despite striking and classic demyelination in the optic nerve and spinal cord, patients with “pure” DNO do not develop neurologic signs or symptoms or demyelination in other regions of the CNS; the brain remains normal or shows at most non-specific findings in the white matter on follow-up MRI (Filippi et al. 1999; Mandler et al. 1993; O’Riordan et al. 1996; Fazekas et al. 1994).
Many series of DNO patients suggest that visual symptoms usually precede spinal symptoms, but the reverse is not uncommon. Severe visual symptoms with blindness may become total and permanent within a few days and bilateral optic neuritis is most common. This contrasts with MS where bilateral optic neuritis is relatively uncommon and the initial visual compromise is usually limited and reversible (Weinshenker 2003). In patients who present with visual system difficulties, transverse myelitis characteristically develops within a few weeks, with severe paraplegia, sensory loss with a distinct level, and sphincter disturbances. Fixed weakness from onset, rather than improvement over time after the initial event, is the typical course. This contrasts with MS where weakness is frequently reversible in the early stages and the spinal cord presentation is that of only partial transverse myelitis. The interval separating the visual and spinal syndromes may be hours, days, or weeks, but in most cases is within 3 months. However,cases with a 2- or more year separation have been described. As no single criterion is specific for DNO, diagnostic criteria have been developed based on multiple factors, including MRI (Wingerchuk and Weinshenker 2003; Wingerchuk et al. 1999; de Seze et al. 2002).

Variants of Multiple Sclerosis |
|
|
|
243 |
|
Table 16.1. Classical features of the MS variants compared to relapsing MS |
|
|
|||
|
|
|
|
|
|
|
Relapsing MS |
Devic’s NMO |
Balo’s concentric scle- |
Schilder’s disease |
Acute (Marburg) |
|
|
|
rosis |
|
MS |
|
|
|
|
|
|
Age |
Childhood to adult |
Childhood to adult |
Childhood to adult |
Predominantly |
Childhood to adult; |
|
|
|
|
children |
typically in young |
|
|
|
|
|
adult |
|
|
|
|
|
|
Typical |
First events often |
Acute onset |
Symptoms suggesting |
Headache, vomit- |
Acute onset, poorly |
course |
subclinical |
|
mass may occur |
ing seizures, visual |
responsive, death |
|
|
Nearly synchronous |
|
problems (cortical |
not uncommon |
|
Early deficits mostly |
myelopathy and |
Typical lesions (one or |
blindness or bilat- |
within weeks or |
|
reversible |
optic neuropathy |
few) at presentation, |
eral optic neuritis) |
months |
|
|
(either first) |
may suggest mass |
|
|
|
Relapsing early, then |
|
|
No prodrome |
Typically mono- |
|
progressive in >50% |
Fixed and severe |
Rarely can occur |
|
phasic |
|
|
deficits with each |
during course of typical |
|
|
|
|
attack |
relapsing MS |
|
|
|
|
|
|
Variable |
|
|
|
Monophasic or |
By MRI more common |
|
|
|
|
relapsing |
and more benign |
|
|
|
|
|
Historically acute, |
|
|
|
|
|
severe but highly vari- |
|
|
|
|
|
able and MS-like |
|
|
|
|
|
|
|
|
MRI |
Brain and cord with |
Brain normala |
Lamellar lesions in iso- |
Large (3×2 cm), |
Multifocal dif- |
|
multifocal lesions, in |
|
lation or accompanying |
bihemispheric |
fuse white matter |
|
brain periventricular |
|
typical MS-like lesions |
brain white matter |
lesions in brain or |
|
>peripheral white |
|
|
lesions may show |
brainstem |
|
|
|
Lamellar pattern at |
edge enhancement |
|
|
Spinal cord short |
Spinal cord swollen, |
autopsy (not expected |
|
|
|
segments (<2 in |
diffuse, transverse, |
in vivo) |
|
|
|
height) |
long vertical pathol- |
|
|
|
|
|
ogy |
|
|
|
|
Partial transverse |
Near full transverse |
|
|
|
|
involvement |
involvement |
|
|
|
|
No T1-hypointensity, |
May show T1- |
|
|
|
|
acute or chronic |
hypointensity (acute |
|
|
|
|
|
and chronic) |
|
|
|
|
|
|
|
|
|
CSF |
OCB, in most |
OCB uncommon |
Insufficient studies |
Normal or not |
OCB may be absent |
|
|
>100 WBC; neutro- |
|
typical for MS |
in acute illness |
|
|
phils common |
|
|
|
|
|
|
|
|
|
Pathology Demyelination |
Demyelination and |
Concentric zones of |
Severe myelin |
with variable, but |
necrosis with severe |
normal myelin alternat- |
loss; large, well |
lesser, axonal injury; |
axonal injury and |
ing with demyelination, |
demarcated, |
lesions of differing |
cavitation; damage |
leading to a mosaic |
bilateral white |
ages often detected |
predominantly or |
pattern of myelin |
matter plaques |
at autopsy; most |
exclusively in optic |
damage in cerebral |
involving cerebral |
old lesions well |
nerve and spinal |
hemispheric white |
hemispheric white |
demarcated from |
cord, often affected |
matter; histology may |
matter, with fewer |
surrounding white |
long segments of |
suggest aberrant remy- |
brainstem, cerebel- |
matter; periven- |
cord |
elination |
lar, and spinal cord |
tricular, subpial, |
|
|
lesions |
gray-white junction |
|
|
|
plaques typical |
|
|
|
Severe acute myelin loss with numerous LFB-positive macrophages; few if any old lesions; most lesions in cerebral hemispheres; lesions may be poorly demarcated due to acute nature
a Brain MRI may reveal non-specific white matter foci. OCB, oligoclonal bands

244 |
J. H. Simon and B.-K. Kleinschmidt-DeMasters |
The clinical course following presentation with DNO is variable. The disorder may be monophasic without recurrent attacks (but with fixed deficits) or the patient may experience multiple severe relapses with step-wise neurologic deterioration (Weinshenker 2003; Wingerchuk and
Weinshenker 2003; Fardet et al. 2003). DNO may also follow an acute progressive and fatal course, with patients suffering respiratory failure and death related to cervical myelitis (Wingerchuk and Weinshenker 2003). Rarely there can be complete clinical recovery without relapse. Prognosis is poor compared to classic MS, with most patients demonstrating severe visual loss or inability to ambulate without assistance within 5 years of onset.
The early literature suggested that DNO was the predominant demyelinating disease in parts of Asia, where MS is relatively rare. There is now evidence from studies in Japan and elsewhere that many cases classified as DNO might better be classified as MS with disproportionate optic and spinal cord involvement. Nevertheless, DNO is still rare in these Asian countries compared to MS, and is even more rare in North America and Europe (Kuroiwa 1985a). An interesting co-association of DNO has been described with autoimmune and connective tissue disease, including systemic lupus erythematosus and Sjögren syndrome (de Seze et al. 2002; O’Riordan et al. 1996; Weinshenker 2003; Cree et al. 2002).
Early and correct diagnosis of DNO is important as current treatment recommendations vary from those for MS (Weinshenker 2003). Treatment of DNO with beta-interferon or glatiramer acetate has been described after an initial misdiagnosis of MS, but azathioprine may be the treatment of choice to suppress attacks (Mandler et al. 1998). DNO may respond to plasma exchange when steroid therapy fails (Keegan et al. 2002). DNO in the presence of anticardiolipin antibodies has been treated with antiplatelet and anticoagulant drugs (Karussis et al. 1998).
16.2.2 Neuropathology
DNO is characterized pathologically by considerably more tissue destruction and loss of axons than is seen in typical MS, with necrotizing demyelination in the spinal cord and optic nerves. The tissue involvement often extends over numerous spinal cord segments. In early cases the cord may be swollen, simulating a tumor, whereas in late stages there may be consid-
erable shrinkage of the cord and cavitation due to the tissue destruction. DNO may show a greater B cell component, more prominent eosinophilic and neutrophilic infiltrates, complement activation, and vascular fibrosis, all rare in typical MS, along with the more MS-like features including T cell infiltrates and the presence of macrophages (Lucchinetti et al. 2002). Depending on how one defines the entity, the remainder of the CNS shows little or no demyelinative disease. The cerebrospinal fluid, in contrast to MS, reveals a pleocytosis of >50 leukocytes, is often neutrophilic, and in most cases there is absence of oligoclonal bands.
16.2.3 Imaging
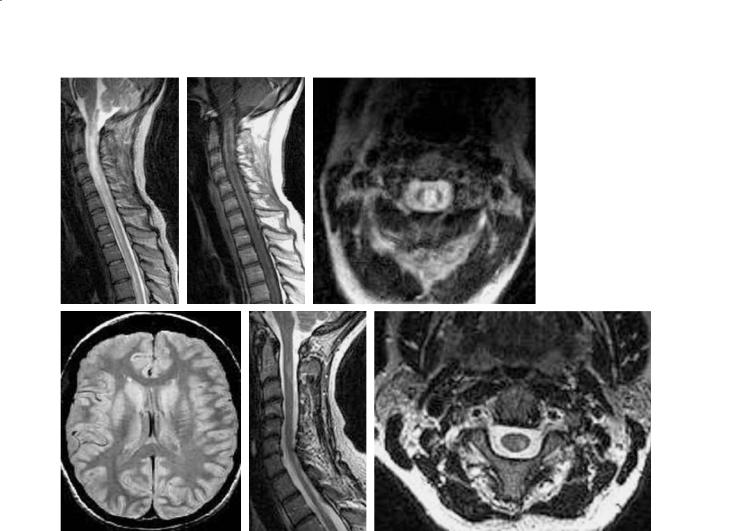
The conventional MRI findings in the spinal cord in DNO differ from those in typical MS (Fig. 16.1). In DNO lesions of the spinal cord tend to be large, often exceeding three segments in height, across the full thickness of spinal cord on axial views. Both the gray and white matter and central cord may be abnormal, and the spinal cord may show striking swelling (O’Riordan et al. 1996; Filippi et al. 1999). This contrasts to the typical appearance of MS by MRI (Lycklama et al. 2003), where there is typically partial and asymmetric involvement observed on axial images, the vertical extent is usually two segments or less, and spinal cord swelling is relatively mild if present at all in the acute stages.
In acute MS the spinal cord is rarely if ever hypointense on T1-weighted images (Gass et al. 1998), while in DNO the cord may be diffusely hypointense. Chronic T1-hypointensity (T1-black holes) are notably absent in MS (Gass et al. 1998; Lycklama et al. 2003), but occur in DNO, probably related to tissue necrosis. In DNO, there may be longitudinally extensive and central enhancement of the spinal cord (Weinshenker 2003).
Cord atrophy does occur frequently in MS, but most often by visual criteria is localized and segmental (Simon 2000; Lycklama et al. 2003). In DNO, cord atrophy may be readily apparent, may be diffuse and prevalent (beyond the acute stages) when measurements are made (Filippi et al. 1999). These neuroimaging changes perfectly parallel the known neuropathological features described above. Imaging findings in the optic nerve or chiasm, including T2hyperintensity, swelling or enhancement, do not distinguish DNO from typical MS.
Variants of Multiple Sclerosis |
245 |
a |
b |
c |
d |
e |
f |
Fig. 16.1a-f. Devic’s neuromyelitis optica. A relapsing spinal cord presentation included an episode with severe long segment, diffuse spinal cord involvement as seen on T2-weighted MRI (a), with corresponding T1-hypointensity (b), findings not associated with classic MS. On axial T2-weighted MRI (c), the cervical cord lesion is large, symmetric, and crosses the midline. The brain MRI in Devic’s neuromyelitis optica is typically normal as shown here (d), but can include non-specific T2-hyperin- tensities. In MS, on T2-weighted sagittal images, the spinal cord lesion is often vertically oriented, but <2 segments in height. Involvement as seen on axial T2-weighted axial images (f) is classically asymmetric, corresponding to the clinical finding of partial transverse myelitis
While there is some variation depending on the criteria used to make the diagnosis of DNO, there is good evidence that the majority of cases will be characterized by a normal brain MRI at presentation (Wingerchuk et al. 1999; Wingerchuk and Weinshenker 2003; Mandler et al. 1993; de Seze et al. 2003; O’Riordan et al. 1996; Fazekas et al. 1994; Filippi et al. 1999). When T2-hyperintensities are seen in the brain, they tend to be non-specific, for example not abutting the ventricular surfaces, and without accompanying T1-hypointensity, either acute or chronic. On follow-up, cases of “pure” DNO do not tend to accumulate new T2-hyperintensities, in contrast to typical MS where new lesions are frequent and most often subclinical (Filippi et al. 1999).
Magnetization transfer imaging studies have provided further support for the concept of DNO as a more limited neuroanatomical disorder in contrast to typical MS. The magnetization transfer ratio (MTR) of focal non-specific brain lesions when they do occur in DNO is only mildly abnormal and considerably less abnormal than in MS. The MTRbased measures of normal-appearing white matter (NAWM) is normal in DNO, in contrast to the NAWM in MS which is consistently abnormal even in the relatively early stages of disease (Filippi et al. 1999). In keeping with a more severe spinal cord pathology, the MTR of spinal cord is significantly more abnormal in DNO than in typical MS (Filippi et al. 1999).

246 |
J. H. Simon and B.-K. Kleinschmidt-DeMasters |
16.2.4
Di erential Diagnosis
Depending on the initial presentation (spinal or optic), the differential diagnosis by imaging includes (for spinal presentations) transverse myelitis, acute disseminated encephalomyelitis (ADEM), including post-vaccination,viral and other infectious etiologies, and neoplasm. An abnormal brain MRI with typical demyelinating lesions in the cerebral hemispheres, cerebellum, and brainstem, and consistent laboratory findings makes MS more likely. Vascular insults and collagen vascular disease are not always separable from DNO without supporting laboratory or historical information. With an optic neuritis presentation, especially if unilateral, imaging findings are non-spe- cific and the differential diagnosis will include optic neuritis from MS if the brain MRI is positive.
16.3
Acute MS (Marburg Type)
16.3.1
General Features
Rarely, an acute, idiopathic, inflammatory demyelinating disease may be relatively unresponsive to conventional therapy (corticosteroids), resulting in death or severe residual deficits, and may then be classified as acute MS of the Marburg type. Death may occur in weeks to months, either from severe widespread cerebral lesions or acute involvement of the lower brainstem or upper cervical cord (Mendez and Pogacar 1988). Patients who survive the acute presentation may be left with significant deficits or may develop severe exacerbations.
It must be acknowledged, however, that some patients who present with a large acute tumefactive demyelinative lesion that prompts biopsy, later go on to exhibit a classic chronic relapsing/remitting course of typical MS. These cases should perhaps not be considered “pure” Marburg type MS although at the time the patient presents the outcome may not be at all clear. For the individual patient, the difficulty is that at the time of clinical presentation there are no good predictors for whether he or she will follow a fulminant rapidly fatal course after biopsy, develop mild or severe MS, or even develop MS at all, despite several years follow-up (Kepes 1993). It has been suggested that some patients who present with acute tumefactive demyelinating lesions prompting biopsy might
actually have disorders intermediate between MS and large coalescent ADEM (Kepes 1993). Hence, the appellation “Marburg” is best applied to severe, acute MS that meets both clinical and pathological criteria of a monophasic illness and may best be defined by its malignant course and acute, severe demyelination (Bitsch et al. 1999; Poser et al. 1992).
Whilepoorlyresponsivetocorticosteroids(thefirst line of therapy), there are suggestions that patients with Marburg-type MS may benefit from plasma exchange (Weinshenker 1999). Cases have also been described with response to combined corticosteroid and mannitol therapy (Giubilei et al. 1997).
Recently, Marburg MS has been hypothesized to be the result of a pre-existing abnormality of myelin basic protein in a developmentally immature (less cationic) form (Wood et al. 1994; Beniac et al. 1999). Alternatively, Marburg type of demyelination may simply lie at the severe, acute end of the clinical spectrum of MS (Coyle 2000), possibly reflecting factors related to a host response.
16.3.2 Neuropathology
In most patients who succumb, demyelinative lesions are usually all of the same acute age. The distribution of lesions is similar to typical MS, but there is a predilection for lesions to occur in the cerebral hemispheres, including near the gray-white matter junction.Virtually no older plaques can be identified at neuropathological examination, paralleling the patient’s rapid clinical downhill course. This is in contradistinction to typical MS where the demyelinative lesions at autopsy are usually remote and/or show differing ages of myelin breakdown. The latter is assessed by several types of special stains, the most common of which is the histochemical stain for myelin (Luxol fast blue, LFB) with a periodic acid Schiff (PAS) counterstain.This stain allows distinction between very recently phagocytosed myelin within macrophages, which is as yet undigested and retains its LFB-positivity, versus myelin that has been broken down to neutral lipids in macrophages (PAS-positivity) as a result of a more advanced and subacute process. In Marburg type of acute MS, abundant LFB-positive macrophages are seen throughout the hypercellular demyelinative lesions. Acute MS shows less chronic gliosis, more edema, and even partial bands of preserved myelin (see description of Balo type in Sect. 16.4) than typical relapsing/remitting MS cases that come to autopsy. More abundant perivascular inflammation may also be present than is seen in typical MS.
Variants of Multiple Sclerosis |
247 |
16.3.3 Imaging
The classic MRI appearance is that of a first presentation with large, often confluent, lesions, sometimes involving the brainstem but more commonly affecting cerebral hemispheric white matter. Lesions may show enhancement and perilesional edema is often present. If a single lesion is the initial presentation of MS, the neuroimaging characteristic may be difficult to distinguish from a neoplasm.
16.3.4
Di erential Diagnosis
The differential diagnosis at the time of presentation includes ADEM (Schwarz et al. 2001), which may be favored by an appropriate accompanying history of vaccination or recent viral or other infectious illness. If blood is present in the lesion at neuroimaging studies, the rare Hurst’s disease (acute hemorrhagic leukoencephalitis) may also be a consideration, a disorder generally considered to represent a hyperacute form of perivenous encephalomyelitis (ADEM). Infectious and autoimmune diseases can usually be excluded by clinical and laboratory features or by neuropathology of the biopsy or at autopsy.
The difficulty with biopsies of tumefactive MS is that they may overlap with Marburg MS or be intermediate between ADEM and classic MS (Giang et al. 1992; Kepes 1993). While classic ADEM is distinguishable from classic MS by the presence of multifocal, small perivenous demyelinative lesions in ADEM, this feature is not generally present in acute demyelinative lesions that prompt biopsy. There are no morphological features in these biopsy specimens of single tumefactive demyelinative lesions that reliably predict whether the patient will go on to subsequently develop MS.
16.4
Balo’s Concentric Sclerosis
16.4.1
General Features
Balo’s concentric sclerosis (BCS) might be best characterized as a relatively rare expression of pathology that might be entirely within the classification of MS. The original case of Balo was that of acute disease,
with death within 3 and a half months after onset in a 23-year-old male, prompting Balo himself to consider the disorder he described as a variant of acute MS; he named this process encephalitis periaxialis concentrica, paralleling the name given several years earlier by Marburg to acute MS (Balo 1928; Kuroiwa 1985a).
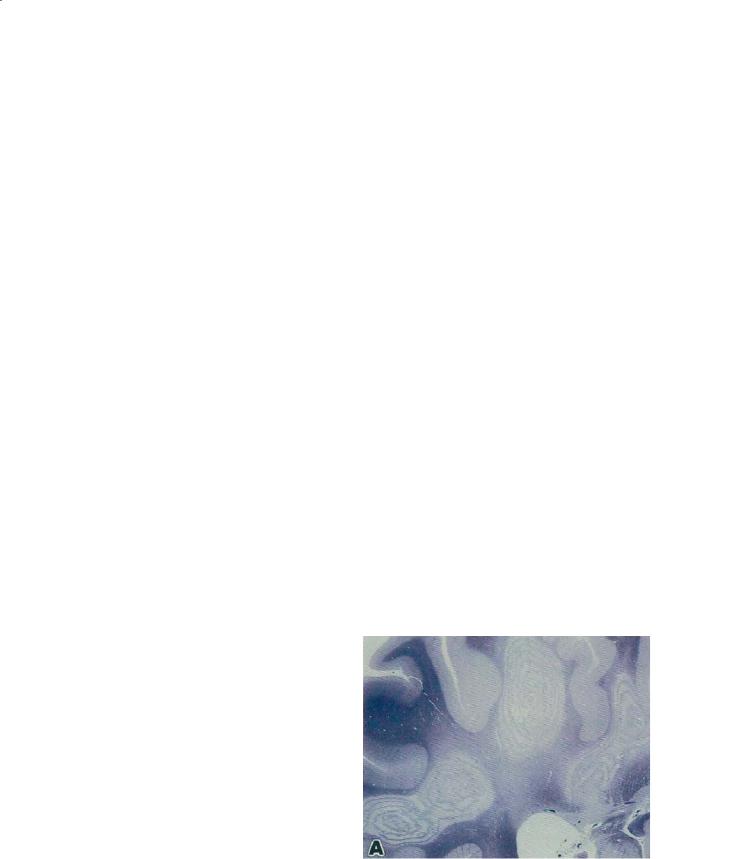
BCS by definition shows a peculiar pattern of pathology in cerebral hemispheric white matter consisting of a concentric, mosaic, or floral configuration of alternating bands of white matter whose basis is relatively preserved myelination alternating with regions of demyelination (Fig. 16.2).
The literature reflects two extremes of BCS – the earlier (pre-MRI) neuropathology literature is based primarily on autopsy material from cases after an acute, monophasic, fulminant disease process. In the autopsy BCS series, disease typically progresses over weeks to months, with severe disability or death as the typical outcome. Clinical symptoms typically include headache, aphasia, cognitive or behavioral dysfunction, and/or seizures.
In contrast, the MRI-era literature tends to reflect a far greater range of disease, from focal BCS lesions co-existing with typical MS-like lesions, but also acknowledging cases with a fulminant course ending in death. The more “benign” BCS has been described as monophasic with resolution of pathology and clinical findings over time, and as MS-like with a multiphasic but self-limited course, and responsive to therapy (Spiegel et al. 1989; Ng et al. 1999; Louboutin and
Elie 1995; Sekijima et al. 1997; Karaarslan et al. 2001).
Fig. 16.2. Classic Balo’s concentric sclerosis as a white matter lesion with partly myelinated and demyelinated bands arranged in concentric and mosaic patterns. Section stained with Luxol fast blue. [From Yao et al. (1994)]

248 |
J. H. Simon and B.-K. Kleinschmidt-DeMasters |
The explanation for the pre-MRI and MRI era dichotomy is not known. One possibility may lie with a more detailed explanation of the pathological features of acute MS. Acute Marburg type MS often shows partial bands of preserved myelin at pathological examination, as explained above. Such cases with limited banding are classified as acute MS by neuropathologists, but might be considered as having “focal or limited Balo rings” based on imaging. Acute tumefactive demyelinative lesions with a few alternating bands of intact and damaged myelin may be the lesions that account for the diagnosis of “Balo rings” by MRI in some patients.
Co-existence of BCS lesions with typical MS-like (non-concentric) lesions has also been recognized in the neuropathology literature (Itoyama et al. 1985; Moore et al. 1985). BCS lesions have been found very rarely to develop after a typical relapsing-remit- ting course of typical MS (Moore et al. 2001), and Balo-like band pattern lesions have been described along the periphery of acute MS plaques (Moore et al. 2001).
While the early literature emphasized BCS within the cerebral hemispheric white matter of the brain, this process also occurs within the optic chiasm and spinal cord, and BCS lesions can occur in the brainstem and cerebellum (Itoyama et al. 1985; Moore et al. 1985).
16.4.2 Neuropathology
While much of the literature of BCS was interpreted as indicating that the abnormal bands (intermingled with normal bands) were the result of partial remyelination, more recent evidence suggests that the abnormal bands are more often areas of early demyelination alternating with preserved myelin (Yao et al. 1994). In some well documented cases there does appear to be remyelination of previously demyelinated fibers (Moore et al. 1985). BCS-like features, without the full blown BCS macroscopic pathology have also been described along the surface of chronic active MS plaques (Moore et al. 2001), suggesting that this pathology may not be characteristic of a separate disease, but reflects a variation in pathology or host response.
The mechanism(s) responsible for this peculiar pathology remain a mystery. Balo postulated a “lecithinolytic enzyme” centrifugally spreading from the center of a lesion (Kuroiwa 1985). A more recent hypothesis is based on downregulation of local de-
myelination by CD8 suppressor T-cells (Moore et al. 2001; Traugott et al. 1983), in both typical MS and BCS lesions, as might occur as well through cytokine action (Moore et al.2001; Canella and Raine 1995). The suppression of demyelination along the surface of a lesion, surrounded by an external zone of activity and demyelination, and a sequential repeat of the process might then result in the repetitive alternating lamellae characteristic of BCS (Moore et al. 2001).
16.4.3 Imaging
With the introduction of MRI, the literature regarding BCS has evolved. BCS is currently described as consisting of a range appearances, from classic large BCS lesions in isolation associated with a fulminant clinical course, to cases in which the BCS pattern of focal lesions co-exists with typical MS-like lesions (Chen et al. 1996; Yao et al. 1994; Iannucci et al. 2000; Ng et al. 1999). There is also increasing recognition of borderline BCS-like lesions with only a few lamellae or rings. The co-existence and development of BCS lesions within typical MS lesions, though rare, is likely more common than development of only classic large BCS lesions throughout both cerebral hemispheres.
BCS lesions are readily identified on proton or T2weighted images, but the concentric pattern may also be apparent on T1-weighted images (Fig. 16.3). There are reports of contrast enhancement in BCS, with alternating bands of enhancement and non-enhance- ment, the enhancing regions thought to correspond to zones of demyelination. Synchronously enhancing, sequentially enhancing, and transiently enhancing rings have been reported (Iannucci et al. 2000; Sekijima et al. 1997; Chen 2001;Chen et al. 1999; Ng et al. 1999; Bolay et al. 1996; Caracciolo et al. 2001; Kastrup et al. 2002) (Fig. 16.4).
By both imaging and neuropathology,most studies suggest a chronologic progression of rings, with the most recent pathology along the periphery. In some cases a more synchronous process has been suggested, possibly associated with a very rapid tempo of lesion progression, not unlike that of chronic active lesions in classic MS, where ongoing demyelination may occur along a lesion’s perimeter (Moore et al. 2001; Sekijima et al. 1997; Ng et al. 1999).
In the few cases of BCS studied to date with MR spectroscopy, the principal metabolite ratios and other abnormal peaks (from lipid and lactate) are those observed from typical large MS lesions
Variants of Multiple Sclerosis |
249 |
a |
b |
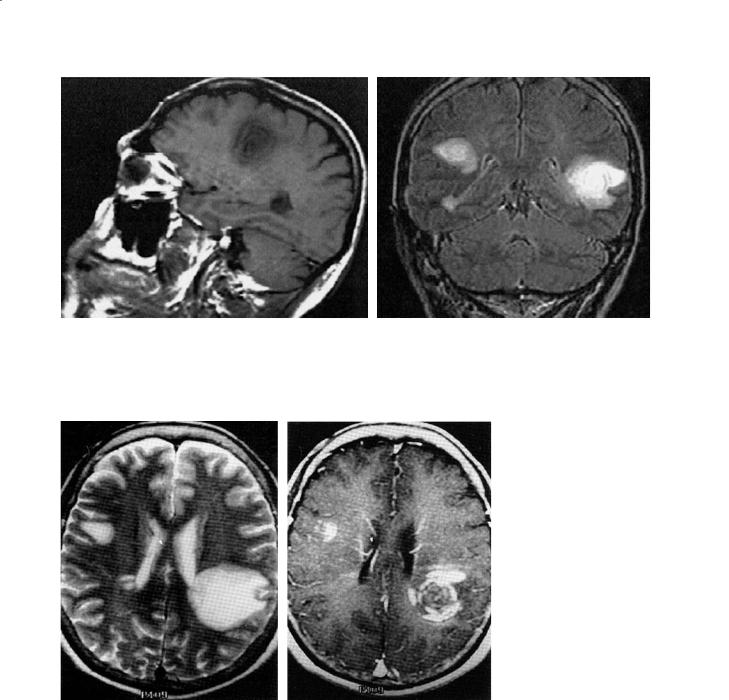
Fig. 16.3a,b. Balo’s concentric sclerosis. (a) A 52-year-old man presenting with acute left hemiparesis, ataxia and agitation. Sagittal T1-weighted image shows concentric rings, the lesion had peripheral enhancement (not shown). (b) A 48-year-old man developed acute sensorial aphasia 4 days before admission, the coronal fluid-attenuated inversion-recovery (FLAIR) series shows bilateral lesions, one with many rings in the left temporo-parietal lobe, and two right hemisphere lesions, one with few rings, one more classically MS-like along the ventricular surface. [From Karaarslan et al. (2001)]
a |
b |
Fig. 16.4a,b. Balo’s concentric sclerosis. A 56-year-old woman with a 2-week history of progressive weakness, the axial T2weighted MRI shows a left parietal and right frontal subcortical lesion, and small right periventricular lesions (a). The axial postcontrast T1-weighted image (b) shows concentric enhancement of the left lesion, arcuate enhancement of the right subcortical lesion, and enhancement at right angle to the ventricle surface in the smaller right periventricular lesion. Follow-up at 12 months showed regression but the laminated appearance remained on T2-weighted imaging (not shown). [From Ng et al. (1999)]
(Karaarslan et al. 2001; Kim et al. 1997; Chen et al. 2001). Based on a series of large concentric lesions in four patients in Taipei, Taiwan, with 2- to 23-month follow-up Chen et al. (2001) reported an increase in the choline-to-creatine ratio, a decrease in the N-ace- tyl aspartate-to-creatine ratio, and increased lactate. On follow-up, there was a return toward normal ra-
tios and values. Lipid peaks were seen in early lesions, similar to those observed in MS by short echo time MR spectroscopy (Davie et al. 1994).
With increased sensitivity of MRI to Balo-like patterns in vivo, one of the difficult and intriguing questions is concerned with the definition of BCS. For example are lesions with only a few or one to two
250 |
J. H. Simon and B.-K. Kleinschmidt-DeMasters |
rings representative of BCS (Fig. 16.5), and its corresponding pathology, or are these better characterized as relatively rare presentations of the common demyelination characteristic of typical MS? Does identification of a minimal BCS pattern contain prognostic information, information relevant to discussions of subtypes of MS pathology or information relevant to optimal therapy?
a
b 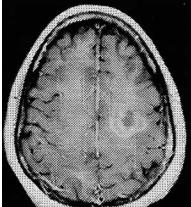
Fig. 16.5a,b. Balo’s concentric sclerosis pattern with few rings. Biopsy-proven inflammatory demyelinating process consistent with Balo’s concentric sclerosis. T2-weighted image (a) shows two concentric zones of abnormal intensity. Post-con- trast T1-weighted image (b) shows multiple concentric rings of enhancement. [From Caracciolo et al. (2001)]
16.4.4
Di erential Diagnosis
Classic large and potentially mass-like BCS lesions may be confused with neoplasm or abscess, but when their appearance is equivocal, the course when followed by imaging should suggest a resolving (non-malignant) process. MR spectroscopy may also support the diagnosis of a demyelinating process (Chen et al. 2001).
16.5
Schilder’s Disease
16.5.1
General Features
Schilder’s disease, also known as diffuse myelinoclastic sclerosis, is a rare demyelinating disorder that is essentially a diagnosis of exclusion as its clinical and MRI appearance may overlap with inherited metabolic disorders of myelin, particularly adrenoleukodystrophy. Schilder’s original three cases, under the umbrella of an entity known as encephalitis periaxialis diffusa, described in 1912, 1913, and 1924 were subsequently found to be three different disorders: myelinoclastic diffuse sclerosis (the 1912 case), adrenoleukodystrophy (the 1913 case), and subacute sclerosing panencephalitis (SSPE) (the 1924 case) (Poser 1985).
A practical definition proposed by Poser (1985) includes the following components: (1) a subacute or chronic myelinoclastic disorder with one or two roughly symmetrical plaques at least 2×3 cm in two of three dimensions; (2) involvement of the centrum semiovale; (3) these being the only lesions based on clinical, paraclinical or imaging findings; (4) adrenoleukodystrophy must be excluded.
Schilder’s disease in its classic sense is acute MS that occurs in childhood.Analogous to other MS variants discussed above, both “pure” forms and “transitional” forms have been described.“Pure” forms predominate in childhood and have plaques confined to the cerebral white matter.“Transitional” forms affected a broader age range of adolescents and adults and show large cerebral plaques combined with more typical MS plaques elsewhere (Prineas et al. 2002).
Disease duration is highly variable. In the 70 cases collected by Poser (1957) the mean duration was 6.2 years, ranging from 3 days to 45 years, but duration was less than 1 year in 40%. The clinical course is diverse, but widespread white matter involvement usually produces subacute or chronic mental and neurological deterioration, spastic paresis, convulsion, and involvement of vision and hearing. Pure psychiatric forms have been described. Increased intracranial pressure, headache, and vomiting may suggest a mass lesion. The cerebrospinal fluid may be normal with only a slight elevation of protein or cell count. Treatment with corticosteroids and cyclophosphamide has been successful in some instances. Cases considered to be Schilder’s disease at onset may follow a downhill progressive course or go on to develop relapsing/remitting disease. As noted above in the discussion of acute MS, it is not possible to deter-

Variants of Multiple Sclerosis |
251 |
mine prognosis. In general, however, early age onset often results in severe neurological deficits.
16.5.2 Neuropathology
The defining feature of Schilder’s disease is sharply demarcated, giant coalescent plaques of demyelination, usually involving the majority of bilateral cerebral hemispheric white matter. Histologically, the features of Schilder’s disease are nearly identical to MS. Using the eponymic designation more broadly to include cases of all ages that show massive bilateral cerebral white matter disease affecting the centrum semiovale, “Schilder’s disease” is not distinguishable from severe MS. Indeed, in a study of 22 cases of MS compared to two of Schilder’s disease, no differences could be found (Gallucci et al. 2001). The histological features, however, that linked Schilder’s first MS case with his subsequent misdiagnosed two further cases (adrenoleukodystrophy and SSPE as noted above), however, was the prominent non-neoplastic lymphocytic cuffing in all three disorders. Even today, these three conditions may be included in the differential diagnosis by neuropathologists for large, inflammatory demyelinating diseases found at biopsy or autopsy. Normal ratios of very long chain fatty acids and absence of involvement of the peripheral nervous system exclude adrenoleukodystrophy.
16.5.3 Imaging
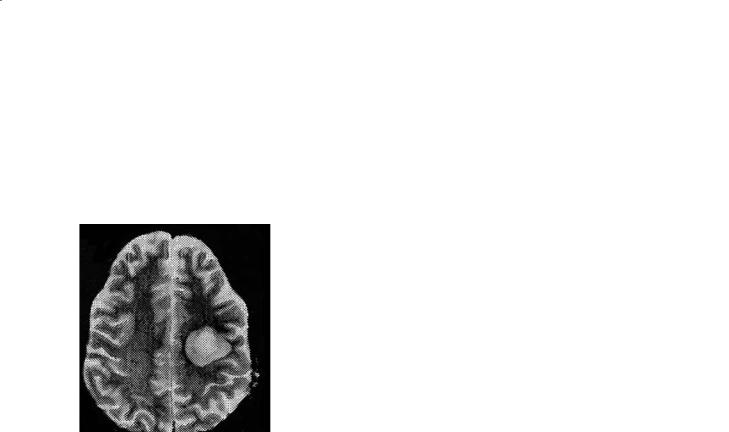
Imaging reveals large, bihemispheric lesions (Eblen et al. 1991; Mehler and Rabinowich 1988) of white matter that may show enhancement at the perimeter (Fig. 16.6). Unfortunately these lesions can also be similar by neuroimaging studies to adrenoleukodystrophy or possibly acute disseminated encephalomyelitis (Fernandez-Jaen et al. 2001;Valk and van der Knaap 1989). The lesions occurring in two hemispheres may be bridged by abnormal signal in the corpus callosum. Frank necrosis and cavitation may also occur.
16.5.4
Di erential Diagnosis
Myelinoclastic diffuse sclerosis remains a rare disorder with few cases meeting rigorous diagnostic criteria (Coyle 2000).Cases coming to biopsy must be distinguished from tumor, abscess, or ADEM (Hynson et al. 2001; Nejat and Eftekhar 2002; Kotil et al. 2002; Kurul et al. 2003) although as noted above, this is often only possible after histological assessment. As expected, the differential diagnosis for Schilder’s disease is very similar to that for other acute forms of MS, such as Marburg type. The additional disorder in the differential diagnosis in children and adolescents is inherited metabolic disorders of demyelination.
a |
b |
Fig. 16.6a,b. Schilder’s disease. A 10-year-old male presented with headache, vomiting, ataxia and foot drop. The MRI reveals multiple large hemispheric (predominantly T2-hyperintense) lesions (a) with ring enhancement (b).Adrenoleukodystrophy was excluded, and neuropathology findings were consistent with Schilder’s disease. (Courtesy of John Strain MD, The Children’s Hospital, Denver)