

2.1.2.1 Метод Хартри-Фока
Метод Хартри- Фока, или метод самосогласованного поля, является одним из эффективных методов решения задач квантовой химии. Его идея состоит в том, что взаимодействие электрона с его окружением заменяется взаимодействием с неким усредненным полем.
В случае нормированной волновой функции Ψ энергию системы можно выразить следующим образом:
![]() (2.1)
(2.1)
![]() (2.2)
(2.2)
 (2.3)
(2.3)
![]() (2.4)
(2.4)
ψp(ξi) - нормированная одноэлектронная волновая функция для i-го электрона, находящегося на p-й орбитали, ξi - обобщенные координаты, включающие в себя пространственную и спиновую часть.
Выражение (2.2) служит отправной точкой для получения уравнений самосогласованного поля Хартри- Фока. Процедура их вывода заключается в том, чтобы минимизировать выражение (2.1) путем варьирования орбиталей, соблюдая при этом требование, чтобы одноэлектронные орбитали были ортонормированы. Система уравнений Хартри-Фока (2.1) решается с помощью метода последовательных приближений, поскольку явный вид оператора Хартри-Фока (кулоновский и обменный потенциалы) зависит от вида искомых волновых функций ψp(ξi). Сущность этого метода состоит в том, что для каждого электрона на первом этапе подбирается пробная волновая функция, с помощью которой рассчитывается оператор Хартри-Фока и решаются уравнения (2.1). Полученные более точные волновые функции снова подставляют в систему уравнений (2.1) и заново ее решают. Этот процесс продолжают до тех пор, пока энергии Ep и вид орбиталей ψp(ξi) на предыдущем и последующем шаге не перестанут различаться на заданную величину. Когда это достигнуто, полученное решение является самосогласованным. По этой причине метод Хартри-Фока называют также методом самосогласованного поля.
Геометрическим образом атомной (молекулярной) орбитали является область пространства вокруг ядра атома (ядер молекулы), где в которой высока вероятность обнаружения электрона. Контуры атомной (молекулярной) орбитали- это графическое отображение поверхности уровня волновой функции, полученной при решении волнового уравнения для одного электрона.
Метод Хартри-Фока не слишком прост при выполнении численных расчетов вследствие сложности обменных слагаемых. Его применение к твердым телам с ковалентной связью приводит, в целом, к правильной форме энергетических зон, но при этом отмечается существенно большее значение ширины запрещенной зоны по сравнению с экспериментом.
2.1.2.2 Метод теории функционала плотности
При численном решении уравнения Шредингера систем с большим числом частиц возникают сложности, связанные с невозможностью вычисления волновой функции с достаточной точностью и записью волновой функции в цифровом виде в память компьютера. Данные проблемы не могут быть решены посредством увеличения точности расчета или расширения памяти, так как в их основе лежит экспоненциальный рост ошибок или памяти. Путь решения проблемы был найден в работах Вальтера Кона с сотрудниками, отмеченных Нобелевской премией по химии 1998г, за разработку метода теории функционала плотности.
Теория функционала плотности (англ. density functional theory, DFT) — метод расчёта электронной структуры систем многих частиц в квантовой физике и квантовой химии. Является одним из наиболее широко используемых и универсальных методов в вычислительной физике и вычислительной химии. В основе метода лежат теоремы Хоэнберга—Кона. Первая теорема утверждает, что существует взаимно однозначное соответствие между плотностью основного состояния электронной подсистемы, находящейся во внешнем потенциале атомных ядер, и самим потенциалом ядер.
Вторая теорема представляет собой вариационный принцип квантовой механики, сформулированный для функционала плотности и утверждает, что энергия электронной подсистемы, записанная как функционал электронной плотности, имеет минимум, равный энергии основного состояния.
Основная цель метода теории функционала плотности- заменить многоэлектронную волновую функцию при описании электронной подсистемы электронной плотностью. Это ведет к существенному упрощению задачи, поскольку многоэлектронная волновая функция зависит от переменных - по 3 пространственных координаты на каждый из электронов, в то время как плотность — функция лишь трёх пространственных координат.
Полную энергию для системы взаимодействующих электронов можно записать в виде:
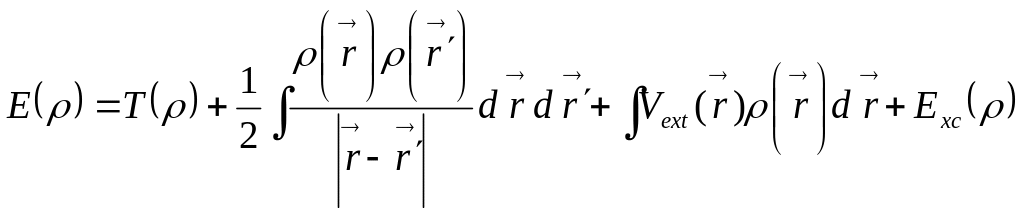 (2.5)
(2.5)
Где Т представляет собой кинетическую энергию, второе слагаемое обеспечивает электростатическое отталкивание электронов. Vext - потенциал взаимодействия электрона с ядрами, а Exс - обменная корреляционная энергия. Вариационное решение этой задачи позволяет получить систему одночастичных уравнений Шредингера вида:
![]()
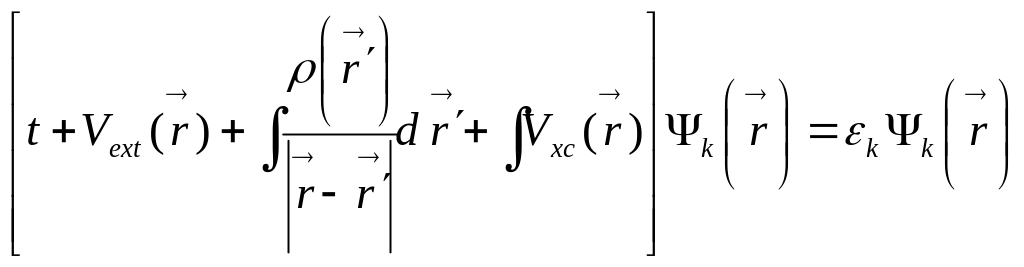 (2.6)
(2.6)
Где t- представляет собой кинетическую энергию отдельного электрона и
![]() (2.7)
(2.7)
Где Vxc(r) - обменно-корреляционный потенциал, включающий все многочастичные взаимодействия.
Основная проблема, связанная с методом теории функционала плотности заключается в том, что точные аналитические выражения для функционалов обменной и корреляционной энергии известны только для частного случая газа свободных электронов. В физических приложениях наиболее распространено приближение локальной плотности, в котором принято, что функционал, вычисляемый для некоторой точки пространства, зависит только от плотности в этой точке:
![]() (2.8)
(2.8)
εxc(ρ) в точке r зависит только от электронной плотности в данной точке ρ(r). Обменная составляющая обменно-корреляционной энергии в приближении LDA определяется формулой Дирака
![]() ,
,
![]()
![]()
Приближение локальной спиновой плотности является непосредственным обобщением приближения локальной плотности, учитывающим спин электрона. Если плотности электронов с разной ориентацией спина α и β не равны, то обменная энергия в приближении LSDA вычисляется по формуле:
![]() (2.9)
(2.9)
Метод обобщённого градиентного приближения (GGA) также является локальным, но, в отличие от метода локальной плотности, учитывает градиент плотности в точке рассмотрения:
![]() (2.10)
(2.10)
Использование этого приближения дает хорошие результаты при расчете геометрии и энергии основного состояния молекул. Существуют и более точные приближения, которые в значительной степени позволяют решить проблему вычисления функционала обменно-корреляционной энергии.
В расчётах квантовой химии одним из распространённых является вид обменного функционала, называемый BLYP (Becke, Lee, Yang, Parr). Еще более широко распространено приближение B3LYP, которое основано на гибридном функционале, в котором обменная энергия рассчитывается с привлечением точного результата, полученного методом Хартри — Фока.