

1.1.2 Редактор GaussView
Графический редактор GaussView позволяет легко конструировать рассчитываемые молекулярные и кластерные структуры в удобном и наглядном для пользователя представлении. Библиотеки редактора GaussView содержат не только атомы, но и молекулы, радикалы, биологические молекулы, существует возможность создавать пользовательские структуры и пополнять ими библиотеку. Все это в сумме позволяет значительно ускорить процесс построения моделируемых структур.
Во-вторых, с использованием редактора GaussView задается используемый расчетный метод. Создание пользовательских схем расчетов (т.е. метод, базис, выделяемый объем памяти и др., используемые по умолчанию) позволяет сократить время на подготовку входного файла (подробнее о настройке параметров при выполнении расчетов будет рассмотрено в лабораторной работе №2). После построения расчетной структуры, установки метода расчета и дополнительных опций редактор генерирует входной Gaussian Job File в формате *.gjf (для операционной системы Windows) или *.com (для Linux). Запуск расчетов в Gaussian можно осуществлять с помощью редактора.
По окончании расчетов в комплексе Gaussian формируется файл, содержащий данные результатов расчетов в формате*.log или *.out. Таким образом, третьей важной задачей редактора GaussView является перевод результатов расчетов в графическое наглядное представление, построение различных графиков. GaussView позволяет визуализировать:
- оптимизированную молекулярную структур, отвечающую минимуму энергии;
- молекулярные орбитали;
- поверхности и контуры электронной плотности;
- заряды атомов и дипольные моменты;
- анимацию вибрационных мод;
- ИК-, ЯМР- и Рамановские спектры;
Внешний вид редактора представлен на рисунке 1.2.
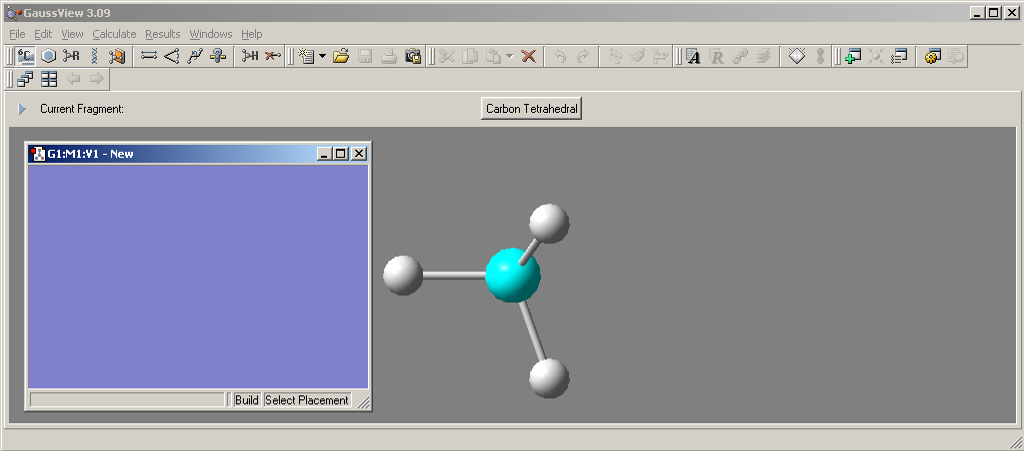
Рис. 1.2. Контрольная панель и активное окно редактора GaussView.
В состав панели меню на контрольной панели редактора входят:
File: создание, открытие и сохранение структур, установка параметров редактора
Edit: построение молекулярных структур
View: управление настройками отображения
Calculate: параметры расчетов и передача входных данных в Gaussian.
Results: просмотр результатов расчетов, включая графики, спектры, поверхности и анимации;
Windows: управление окнами GaussView.
Табл.1.1. Панель команд редактора GaussView
|
|||
|
библиотека элементов Element Fragment |
|
изменение длины и типа химических связей Modify Bond |
|
библиотека циклических соединений Ring Fragment |
|
изменение величины валентных углов Modify Angle |
|
библиотека радикалов R-Group Fragment |
|
изменение величины дигедральных углов Modify Dihedral |
|
библиотека биомолекул Biological Fragment |
|
увеличение валентности Add Valence |
|
база пользователя Custom Fragment |
|
удалить атом Delete Atom |
|
|||
|
численное задание координат Atom List Editor |
|
кристаллический редактор PBC Editor |
|
дополнительные параметры координат Redundant Coordinate Editor |
|
редактор молекулярных орбиталей MO Editor |
|
|||
|
вырезать выделенное |
|
отменить последнее действие |
|
копировать выделенное |
|
перерасчет длин связей |
|
вставить из буфера обмена |
|
проверка геометрии на соответствие правилам |
|
удалить выделенное |
|
определение точечной группы симметрии |










Табл.1.2 Использование мыши в редакторе GaussView
функция |
действие мышью в рабочем окне |
выделение или вставка атома |
нажать левую кнопки |
вращение всех объектов |
зажать левую кнопку |
вращение выделенной молекулы |
зажать Alt+ левая кнопка |
вращение в плоскости рабочего окна |
зажать Alt+ правая кнопка/ Ctrl+(Left, Right) |
перемещение |
зажать Shift+левая кнопка/ средняя кнопка/Shift+(Left, Right, Up, Down) |
перемещение выделенной молекулы |
зажать Alt+ средняя кнопка |
вызов контекстного меню |
нажатие правой кнопки |
приблизить/удалить |
зажать Ctrl+левая кнопка/ правая кнопка/ Ctrl+(Up, Down) |