

3 курс / Патологическая анатомия / Частная патанатомия
.pdf231
тромбозы межворсинчатых пространств и др.) с различными вариантами дисэмбриогенеза ворсинчатого хориона.
Острая плацентарная недостаточность чаще приводит к мертворожденности, хроническая – к гипотрофии, незрелости, асфиксии недоношенного и новорожденного.

232
РАЗДЕЛ X ПРЕНАТАЛЬНАЯ ПАТОЛОГИЯ
Пренатальная (антенатальная) патология включает в себя все патологические процессы и состояния человеческого зародыша от момента оплодотворения и до рождения ребенка.
Пренатальный период у человека составляет 280 дней или 40 недель.
Все развитие, начиная от созревания половой клетки (гаметы) до рождения зрелого плода, делят на два периода:
период прогенеза – соответствует созреванию гамет (яйцеклеток и сперматозоидов) до оплодотворения и называется также гаметогенез. В этот период возможно возникновение патологии гамет (гаметопатий);
период киматогенеза – соответствует периоду от оплодотворения до родов:
бластогенез – период от оплодотворения до 15 дня беременности. В этот период идет дробление яйца, заканчивается образованием эмбриобласта и трофобласта
(патология – бластопатии);
эмбриогенез – период с 16 дня до 75 дня беременности, идет основной органогенез и образуется амнион и хорион (патология
– эмбриопатии);
фетогенез – период с 76 дня по 280 день беременности, происходит дифференцировка и созревание тканей плода, образование плаценты, а также рождение плода (патология – фетопатии). В свою очередь фетогенез делят на:
o ранний фетальный период (76-180 день беременности); o поздний фетальный период (181 -280 день беременности).
ЭТИОПАТОГЕНЕЗ
В большинстве случаев этиопатогенез развития пренатальной патологии неясен. Определенное значение имеет ряд факторов:
вирусы краснухи, кори, герпеса, гепатита, паротита, полиомиелита, гриппа и др.;
ионизирующее излучение;
лекарственные препараты (гормоны и цитостатики);
эндокринные заболевания матери (сахарный диабет, болезни щитовидной железы);
алкогольная и никотиновая интоксикация (алкогольная и никотиновая эмбриофетопатия) и др.

233
Впатогенезе имеются некоторые закономерности:
повреждение того или иного органа или системы связано со временем действия на плод этиологических факторов
(тератогенный терминационный период – отрезок времени в ходе киматогенеза, в течение которого тератогенный фактор способен вызывать формирование порока развития органа.);
любое патогенное воздействие в ходе морфогенеза приводит к изменениям при формировании органов и систем;
на поздних этапах развития наряду с нарушением дифференцировки в органах и тканях плода развиваются общепатологические процессы – некроз, кровоизлияния, воспаление, гипо- и гипертрофия.
►ГАМЕТОПАТИИ◄
Гаметопатии – повреждения мужской и женской гаметы (яйцеклетки или сперматозоида), возникающие во время овоили сперматогенеза до оплодотворения.
Наибольшее значение имеют дефекты генов гамет (генные мутации) и хромосом (хромосомные мутации или хромосомные аберрации), приводящие к развитию наследственных заболеваний –
генных и хромосомных болезней соответственно.
ГЕННЫЕ МУТАЦИИ
Генетические (генные) заболевания подразделяются на 4 типа в зависимости от типа наследования:
аутосомно-рецессивные (родители при этом могут быть здоровыми, являясь гетерозиготными носителями дефектного аллеля); примеры: болезни накопления (болезнь Гоше, болезнь Ниманна-Пика и др.);
аутосомно-доминантные (родители также страдают этим заболеванием); примеры: семейная гиперхолестеринемия, синдром Марфана и др.
рецессивные, сцепленные с Х-хромосомой (встречаются обычно у мальчиков) – мутантный ген ребенок получает от матери, которая является носительницей дефектного гена и не болеет; примеры: дальтонизм, гемофилия тип А и В, др.
доминантные, сцепленные с Х-хромосомой; примеры: гемофилия тип С, мышечная дистрофия Дюшенна-Беккера и др.
234
ХРОМОСОМНЫЕ МУТАЦИИ
Хромосомные болезни – наследственные заболевания, обусловленные изменением числа или структуры хромосом. К хромосомным болезням относят патологические состояния, обусловленные геномными мутациями или структурными изменениями отдельных хромосом.
Типичными примерами хромосомных болезней являются болезнь Дауна, синдром Патау, синдром Клайнфельтера, синдром Шерешевского-Тернера, синдром Эдвардса. Ниже представлена краткая характеристика этих синдромов.
Болезнь Дауна – трисомия по 21 паре хромосом (47), которая клинически проявляется задержкой умственного и физического развития. Частота возникновения болезни Дауна увеличивается с возрастом матери.
Характерным является внешний вид такого больного:
брахицефалия (низкий лоб и скошенный затылок);
косой разрез глаз;
эпикант (вертикальная складка кожи полулунной формы, прикрывающая внутренний угол глазной щели);
западение спинки носа;
низкие маленькие ушные раковины;
гипотония мышц;
патогномоничный признак – пятна Брушвильда (участки депигментации радужки);
поперечная ладонная складка.
Обычно, причиной смерти в таких случаях является хроническая сердечная недостаточность на фоне порока развития сердечнососудистой системы (чаще септальные дефекты и тетрада Фалло) и лейкоз.
Синдром Патау – трисомия по 13 паре хромосом (47). Помимо отставания в умственном и физическом развитии у таких детей имеется:
низкий скошенный лоб;
расщелины верхней губы и твердого неба;
полидактилия и флексорное положение кистей;
анофтальмия или микрофтальмия и помутнение роговицы;
микроцефалия, аринэнцефалия;
пороки развития внутренних органов.
патогномоничный признак – увеличение дольчатости почек и поликистоз;
эктопия ткани селезенки в поджелудочную железу;
удвоение матки и влагалища.

235
Синдром Клайнфельтера – кариотип 47 ХО или ХХО (ХХХО, ХХХХО
ит.д.).
диагностируется, как правило, после полового созревания;
высокий рост за счет длинных конечностей;
трудности в учебе, редко олигофрения (25-30%);
недоразвитие половых желез и бесплодие;
гинекомастия;
остеопороз;
ожирение;
сахарный диабет 2 типа.
Синдром Шерешевского-Тернера – моносомия 45 ХО.
низкорослость;
инфантильное строение половых органов, аменорея, стерильность;
"бочкообразная грудная клетка"
короткая шея с "крыловидными" складками;
деформации ушных раковин и локтевых суставов;
интеллект у этих детей не страдает, живут они долго.
Синдром Эдвардса – трисомия 18 пары (47).
долихоцефалия со ступенеобразным черепом;
микрогирия (истончение, укорочение и увеличение количества извилин больших полушарий головного мозга);
флексорное положение кистей;
"стопа-качалка";
синдактилия;
гипоплазия мозжечка;
патогномоничный признак – утолщение дорсальных зубчатых ядер оливы.
►БЛАСТОПАТИИ◄
Бластопатии – киматопатии, возникающие в периоде бластогенеза (1-15 день антенатального развития).
В период бластогенеза происходит дробление зиготы и образование тканевых зачатков. Различают следующие наиболее распространённые варианты бластопатий:

236
патология имплантации бластулы (эктопическая беременность,
поверхностная или глубокая имплантация бластулы в эндометрий). Поверхностная имплантация чаще приводит к нарушению расположения плаценты и самопроизвольным абортам, глубокая ведёт к её приращению. Нарушение локализации имплантации влечет за собой развитие внематочной беременности, а глубокая имплантация способствует приращению плаценты;
образование пустого зародышевого мешка вследствие аплазии или гибели эмбриобласта;
аплазия/гипоплазия трофобласта;
панэмбриональные аномалии (несовместимы с жизнью);
пороки развития отдельных органов, множественные или одиночные (в половине случаев они сочетаются с пороками развития провизорных органов);
двойниковые уродства (сросшиеся двойни) – развиваются при наличии общей зоны между двумя центрами дробления. Если двойня состоит из равно развитых компонентов, то ее называют "диплопагусом", если один из близнецов разит меньше чем второй, то двойня называется "гетеропагусом", а меньший близнец – паразитом. Для обозначения локализации сращения к анатомическому образованию места сращения добавляют слово "пагус" (например, краниопагус – сращение в области головы, торакопагус – сращение в области грудной клетки, ишиопагус – сращение в области таза). Двойниковые уродства, как правило, сочетаются с нежизнеспособностью.
►ЭМБРИОПАТИИ◄
Эмбриопатии – киматопатии, возникающие в периоде эмбриогенеза (16-75 дни внутриутробной жизни).
В период эмбриогенеза нарушения связаны с развитием врожденных пороков развития, под которыми понимают стойкое морфологическое изменение органа, части тела или всего организма, выходящее за пределы вариаций нормального строения определенного биологического вида, возникающее в результате нарушения морфогенеза.
Кроме нарушения анатомического строения возможно развитие нарушения на тканевом уровне – пороки развития поперечно-полосатой мускулатуры, соединительной ткани, кожи, костей хрящевого генеза и др. Классификация врожденных пороков развития представлена ниже
(см. рис. 46).
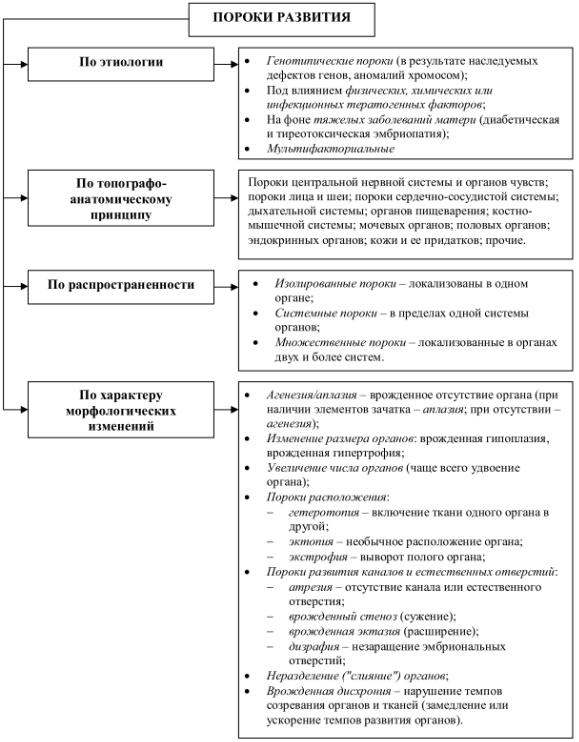
237
Рис. 46. Классификация врожденных пороков развития.
Ниже представлена краткая характеристика основных пороков развития по системам органов.
ВРОЖДЕННЫЕ ПОРОКИ СЕРДЦА
Врожденные пороки сердца и сосудов представляют собой один из наиболее распространенных видов пороков, уступая по частоте лишь

238
порокам развития центральной нервной системы.
Врожденные пороки сердца могут наблюдаться или в камерах сердца, или в крупных сосудах. Наиболее частые пороки классифицируются на основе:
структуры поражения сердца;
наличия сообщения или шунтирования между двумя половинами;
для пороков со сбросом крови по наличию или отсутствию цианоза (условно, не всегда применимо):
пороки "синего типа" (с право-левым сбросом крови, со смешиванием артериальной и венозной крови) – наблюдается цианоз, который возникает в результате сброса крови справа налево, вследствие чего неоксигенированная венозная кровь, минуя легкие, попадает в большой круг кровообращения (центральный цианоз);
пороки "белого" типа (бледные, с лево-правым сбросом крови, без смешивания артериальной и венозной крови).
Врожденные пороки сердца, преимущественно, "белого типа"
Незаращение боталлова (артериального)
протока – кровь из аорты поступает через открытый артериальный проток в легочную артерию. При легочной гипертензии – наоборот (в таком случае – это порок "синего" типа).
Дефект межжелудочковой перегородки –
врожденный порок сердца, характеризующийся наличием дефекта между правым и левым желудочками сердца. Гемодинамические расстройства выражаются в сбросе крови через дефект слева направо. При наличии легочной гипертензии ток крови будет обратный (порок "синего" типа).

239
Дефект межпредсердной перегородки –
врожденный порок сердца, характеризующийся наличием дефекта между правым и левым предсердиями сердца. Сброс крови слева направо. При наличии легочной гипертензии ток крови будет обратный (порок "синего" типа).
Общий артериальный ствол, отходящий от обоих желудочков. Синий тип порока. Часто при этом пороке дети оказываются нежизнеспособны.
Полная транспозиция аорты и легочной артерии. Кровоток в большом и малом кругах кровообращения полностью разобщаются, оксигенированная кровь может попасть в большой круг кровообращения только через дефекты перегородок или незаращенный Боталлов проток. Дети нежизнеспособны.
Стеноз и атрезии легочного ствола или аорты. Жизнеспособность и прогноз в зависимости от степени стеноза и степени нарушения кровообращения.
Врожденные пороки сердца, преимущественно, "синего типа"
Триада Фалло – дефект межпредсердной перегородки, стеноз легочной артерии, гипертрофия правого желудочка.
Гемодинамика: из-за повышенного давления в правых отделах сердца (в результате стеноза легочной артерии) кровь через отверстие в межпредсердной перегородке поступает в левое предсердие, минуя легкие.
Тетрада Фалло – дефект межжелудочковой перегородки, стеноз устья легочной артерии, гипертрофия правого желудочка и декстрапозиция аорты. Гемодинамика: значительная часть венозной крови сбрасывается из правого желудочка непосредственно в аорту, где она подмешивается к артериальной крови.
Пентада Фалло – аномалии, характерные для тетрады Фалло + дефект межпредсердной перегородки. Гемодинамические нарушения такие же. При небольшом стенозе легочной артерии давление в правом желудочке невелико и оказывается ниже, чем в левом желудочке,

240
поэтому кровь поступает из левого желудочка в правый ("белый тип" порока).
Атрезия трехстворчатого клапана + дефект межпредсердной перегородки. Основным анатомическим признаком данного порока является отсутствие сообщения между правым предсердием и правым желудочком. Гемодинамика: венозная кровь из правого предсердия поступает в левое предсердие и там смешивается с артериальной кровью. В левом желудочке часть крови идет в аорту, а другая часть – в правый желудочек.
Атрезия (отсутствие) правого желудочка. Гемодинамика: венозная кровь из правого желудочка попадает в левый и смешивается с артериальной кровью. Из левого желудочка кровь идет в аорту, из аорты часть крови попадает через Боталлов проток в легочную артерию и в легкие.
ПОРОКИ РАЗВИТИЯ ЦНС
Возникают наиболее часто, в их этиологии наибольшую роль играют вирусы и лекарственные препараты.
Анэнцефалия – отсутствие передних, средних или задних отделов головного мозга. Продолговатый и спинной мозг сохранены;
акрания – отсутствие костей мозгового черепа – часто сочетается с анэнцефалией;
микроцефалия – гипоплазия головного мозга;
микрогирия – уменьшение величины мозговых извилин с увеличением их количества;
порэнцефалия – кистообразование в головном мозге;
гидроцефалия – накопление ликвора в желудочках мозга (внутренняя) или в субарахноидальном пространстве (наружная). В обоих случаях наступает атрофия головного мозга в результате сдавления, а размеры головки плода увеличиваются;
циклопия – наличие одного или двух глазных яблок в одной глазнице. Обычно сочетается с пороком развития носа и обонятельного мозга;
грыжи головного и спинного мозга: наличие в грыжевом мешке оболочек мозга – менингоцеле; наличие в грыжевом мешке оболочек и вещества мозга – менингоэнцефалоцеле; наличие в грыжевом мешке оболочек, вещества и желудочков мозга – менингоэнцефалоцистоцеле. Грыжи спинного мозга часто связаны с расщеплением дорсальных отделов дуг позвонков (spina bifida).