

3 курс / Патологическая физиология / Атеросклероз_сосудов_сердца_и_головного_мозга
.pdf120 Глава 4. ПАТОГЕНЕЗ, ПАТОФИЗИОЛОГИЯ И КЛИНИКА КОРОНАРНОГО И КАРОТИДНОГО АТЕРОСКЛЕРОЗА
зом, располагаются в определенных отделах мозга в соответствии с локализацией и распространенностью атеросклеротического поражения сосудов. Разумеется, возможны и обратные взаимоотношения [207].
Наиболее часто встречающейся формой патологии экстракраниальных отделов ВСА и позвоночных артерий является атеростеноз, который чаще всего определяется у лиц в возрасте 40–60 лет, причем значительно чаще у мужчин. Подчеркивая важность патологии ВСА в развитии ишемии мозга, некоторые авторы придают ей даже черты нозологической определенности, обозначая термином «болезнь внутренней каротиды» (Internal Corotid Artery Disease) [226]. При этом атеросклеротическое поражение синокаротидной области имеет свои особенности и проявления, на которые указывал еще А.Л. Мясников [33].
Видимо, надо напомнить читателю, что каротидный синус расположен в передне-боковом отделе шеи на уровне щитовидного хряща в углу, образованном передним краем грудино-ключично-сосцевидной мышцы и гортанью, и представляет собой колбовидное расширение общей сонной артерии на месте бифуркации ее на внутреннюю и наружную сонные артерии (с обеих сторон). Рецепторы каротидного синуса, расположенные между мышечными пучками сосудистой стенки, раздражаются при ее растяжении, что происходит при общем
èместном повышении АД. Регуляторное действие каротидного синуса через мозговой ствол, симпатический
èпарасимпатический отделы вегетативной нервной системы играет большую роль в поддержании равновесия деятельности сердца, состояния просвета артериального русла, уровня АД и адекватного кровоснабжения мозга. При снижении давления в каротидном синусе увеличивается ЧСС; повышение давления внутри каротидного синуса вызывает брадикардию, расширение периферического сосудистого русла, снижение АД и другие признаки возбуждения парасимпатической нервной системы.
Имеется предположение, что атеросклероз синокаротидной зоны, нарушая механические свойства стенки сонной артерии и кровоснабжение каротидных рецепторов, может вызывать дисфункцию регуляторной активности каротидного синуса [227] и снижать возбудимость хеморецепторов этой сосудистой рефлексогенной зоны [228].
Клинический синдром нарушенной чувствительности каротидного синуса, или синдром каротидного синуса характеризуется триадой: брадикардия, гипотония, обморок. Соответственно выделяют три формы синдрома – кардиально-ингибиторный, вазодепрессорный и церебральный.
Óбольных с гемодинамически невыраженным атеросклерозом сонных артерий выявлено снижение синокаротидного барорефлекторного контроля сердечно-со- судистой системы. Одним из неблагоприятных последствий подобного ухудшения является ослабление демп-
фирующей функции такого рода контроля и увеличение суточной вариабельности уровня АД [229]. Не претендуя на ключевую роль, этот патогенетический механизм может рассматриваться как один из элементов той связи, которая потенцирует взаимоотягощение атеросклероза и артериальной гипертонии. Здесь же надо добавить, что в одном из популяционных исследований установлено, что атеросклероз сонных артерий быстрее прогрессирует у мужчин с гипертензивной реакцией на стресс и высокими психологическими нагрузками на работе [230].
У пожилых людей изолированное повышение систолического АД объясняют снижением податливости аорты и крупных артерий, причинами которого может быть потеря эластичности мышечных волокон, отложение коллагена и кальция в стенке сосудов, исчезновение сосудистых бета-рецепторов [231]. Но А.Л. Мясников главное внимание в патогенезе атеросклеротической артериальной гипертензии уделял цереброишемической концепции. Он так описал возможный патогенез артериальной гипертонии при атеросклерозе: «Один из механизмов развития такой гипертонии состоит в нарушении функции того отдела мозга, в котором заложены вазомоторные центры… Не подлежит сомнению тот факт, что при нарушениях кровоснабжения и питания коры головного мозга, в последней легче и в более резкой степени нарушаются процессы возбуждения и торможения, что приводит к усилению наклонности и неадекватным реакциям раздражения, идущим с интерорецепторов, а следовательно, к невротическим состояниям. Понятно, что подобные условия должны способствовать развитию гипертонической болезни. Поэтому надо считать вероятным, что в результате атеросклероза мозговых сосудов может возникнуть гипертоническая болезнь» [232]. Заметим, что дальнейшие исследования подтвердили полную обоснованность взглядов А.Л. Мясникова на гипертонию у пожилых людей как на феномен, связанный с развитием атеросклероза, прежде всего МАГ, и полагать в основе некоторых форм атеросклеротической артериальной гипертонии цереброишемические механизмы [231, 233].
Роль атеросклеротического поражения экстракраниальных отделов МАГ в развитии различных форм ишемических НМК была установлена в 60–80-е годы. Большой вклад в их описание внесли отечественные ученые Е.В. Шмидт, Н.В. Верещагин, А.Н. Колтовер, А.В. Покровский, Е.И. Гусев, В.А. Хилько и другие.
Нарушение кровотока в системе ВСА атеросклеротического генеза характеризуются широким спектром морфологических изменений мозга и клинических проявлений. Это связано с тем, что через каротидную систему в норме мозг получает до 70% общего количества притекающей к мозгу крови, а также с большим объемом тех отделов мозга, которые получают кровь через систему ВСА [207]. Морфологические изменения, обнаруживаемые при атеросклеротическом поражении
4.2. Ишемическая болезнь головного мозга |
121 |
этой системы, весьма разнообразны как в качественном, так и в количественном отношении. Они отражают прежде всего тяжесть и распространенность ишемии мозга в каротидной системе, ее давность, формы проявления и исход.
Е.В. Шмидт подчеркивал, что сужение и закупорка сонной артерии в одном и том же месте могут давать в разных случаях совершенно различную картину – от тяжелых и стойких расстройств до легких, мимолетных нарушений и могут даже протекать совершенно латентно, обнаруживаясь совершенно случайно на вскрытиях. Выраженность клинических проявлений зависит больше от состояния систем, обеспечивающих коллатеральное кровообращение, чем от степени сужения самой сонной артерии [212].
Стенозы в экстракраниальном отделе сонной артерии почти с одинаковой частотой выявляются как слева, так и справа. Наиболее часто пораженными отделами ее являются область бифуркации общей сонной артерии и синус ВСА. Изменения стенотического характера в других участках цервикального отдела сонной артерии обнаруживаются реже.
Атеросклеротическая бляшка в области бифуркации общей сонной артерии может одновременно приводить к стенозу и внутренней, и наружной сонной артерии, что бесспорно влияет на гемодинамику, ухудшая коллатеральное обеспечение по экстракраниальным анастомозам.
В суженных участках ВСА часто наблюдается пролиферация клеток внутренней оболочки сосудов. Это обусловлено влиянием увеличенной линейной скоростью кровотока и снижением давления в стенозированных участках. В стенке артерии проксимальнее стеноза нередко развивается гиперэластоз, что также, по-види- мому, связано с особенностями гемодинамики в этом участке артерии.
Определение степени выраженности атеростеноза в экстракраниальных отделах ВСА имеет исключительно важное значение для оценки роли этой патологии в нарушениях церебральной гемодинамики. Степень сужения ВСА ранее условно подразделяли по показателям: 50% просвета сосуда, а также менее и более этой вели- чины. В настоящее время гемодинамически и патогенетически значимым считается стеноз ВСА на 70% и более. При этом могут появляться количественные и каче- ственные изменения кровотока, приводящие к недостаточному кровоснабжению мозга. Возникает состояние неустойчивого потока крови, турбулентное движение потока, уменьшается объемный кровоток, который не компенсируется за счет усиления сердечной деятельности [207].
Установлена роль степени каротидного стеноза в качестве основного критерия, позволяющего определить степень риска развития инсульта. Во многих исследованиях показано, что с увеличением степени стеноза возрастает риск развития транзиторных ишемических атак,
эпизодов монокулярной слепоты, а также более стойкого неврологического дефицита [226].
Атеросклеротические бляшки в синусах ВСА всегда сегментарны. Как правило, они имеют веретенообразную форму, реже циркулярную. По своей структуре они не отличаются от бляшек в других отделах артериальной системы человека и имеют в основном слоистое строение. Поверхностный слой грубоволокнистый, в глубине бляшки определяются атероматозный детрит с кристаллами холестерина и петрификатами, а также липофаги, сидерофаги, значительное количество тонкостенных новообразованных сосудов, лимфоцитарные инфильтраты, моноциты, иногда эозинофилы. Новообразованные сосуды являются источником кровоизлияний в бляшку, сопровождающихся отеком бляшки, что приводит к быстрому увеличению объема бляшки и соответственно степени стеноза или даже к закрытию просвета артерии.
Морфологические особенности бляшек, которые имеют значение в повышении риска развития инсульта, могут быть сгруппированы следующим образом [207, 234]:
1)величина – степень вызванного ею стеноза;
2)конфигурация поверхности – гладкая, шероховатая, изъязвленная;
3)гистологическая структура – отложения липидов и атероматозных масс, фиброз, кальцификация, интрамуральные геморрагии.
Сейчас особое внимание уделяется такому компоненту, как кровоизлияние в бляшку, которое рассматривается в качестве маркера повышенного риска НМК. Показано, что выраженные стенозы, обнаруженные у 80% «симптомных» больных, были связаны не только с фиброзом бляшек, но и с внутрибляшечным кровоизлиянием, которое приводит к увеличению бляшек. При этом «стабильная» бляшка может стать «нестабильной» [235]. Кровоизлияния чаще локализуются в глубоких слоях бляшки, в области многочисленных новообразованных сосудов. Показано, что как поверхностные, так и глубоко расположенные новообразованные сосуды имеют очень тонкие стенки; их разрыв приводит к кровоизлиянию в бляшку, что сопровождается увеличением ее объема и, соответственно, степени стеноза ВСА [207].
Изъязвление бляшки может привести к поступлению эмбологенного материала (атероматозные и обызвествленные массы, кристаллы холестерина) в просвет сосуда и попаданию его с кровотоком в ветви ВСА, что приводит к развитию инсульта или транзиторных ишеми- ческих атак. Нередко при изъязвлении формируется пристеночный или обтурирующий тромб, которые также могут быть эмбологенными и приводить к острым НМК. При нарушении фиброзной капсулы кровь может проникнуть во внутренние слои бляшки, а нередко приводить к расслоению артерии или ее тромбозу. Эти про-
Рекомендовано к изучению сайтом МедУнивер - https://meduniver.com/

122 Глава 4. ПАТОГЕНЕЗ, ПАТОФИЗИОЛОГИЯ И КЛИНИКА КОРОНАРНОГО И КАРОТИДНОГО АТЕРОСКЛЕРОЗА
цессы могут трансформировать «асимптомную» бляшку в «симптомную» или, другими словами, «стабильную» – в «нестабильную» [236]. По многочисленным литературным данным подчеркивается, что наиболее высокий (70–75%) риск развития инсульта связан с тромбозом, стенозом артерии, негомогенной структурой бляшки, обусловленной кровоизлиянием в нее, изъязвлением бляшки. Ведущая роль по распространенности и частоте НМК отводится гемодинамически значимому стенозу ВСА [207, 234–236]. При этом отмечена зависимость между выраженностью артериального стеноза и частотой встречаемости того или иного типа атеросклеротической бляшки. Так, бляшки с изъязвлением наиболее распространены при стенозе артерии на 60–80%, бляшки с геморрагическим очагом – при стенозе более чем на 80% [206].
Сужение ВСА вызывает изменения местной гемодинамики, которые могут приводить к реализации двух основных патогенетических механизмов НМК при патологии МАГ – артерио-артериальных эмболий и мозговой сосудистой недостаточности. По данным многих исследователей, изучавших патомеханику кровотока при стенозах сосудов [72, 76, 218, 237], в зоне стенозирования возникают гемодинамические феномены, такие, как отрыв пограничного слоя, повышение напряжения сдвига, формирование области турбулентного кровотока, что способствует как пристеночному тромбозу, так и созданию условий для отрыва тромботических масс и реализации, таким образом, механизма артерио-артериальной тромбоэмболии, в частности, сосудов мозга.
С другой стороны, стеноз ВСА является гемодинамическим барьером, который при определенных условиях может привести к падению объемной скорости кровотока в артерии и возникновению сосудистой мозговой недостаточности. К таким условиям относят прежде всего степень сужения сосуда, состояние остальных МАГ, особенности центральной гемодинамики, активность механизмов ауторегуляции мозгового кровообращения; определенную роль могут играть форма атеросклеротической бляшки, ее протяженность и характер поверхности.
Особенностью септального стеноза с гемодинами- ческой точки зрения является то, что он не только суживает просвет сосуда, но и выполняет роль своеобразного клапана, временно перекрывающего сосуд при изменении локальной или системной гемодинамики и вызывающего развитие НМК. Кроме того, изменения гемодинамики в зоне септального стеноза могут способствовать и тромбогенезу, создавая условия для развития тромбоэмболии [237].
Среди гемодинамических факторов, играющих определенную патогенетическую роль в разные сроки ишемических НМК, в настоящее время рассматриваются свойства потока крови.
Заслуживает внимания тот факт, что в нормальной крови человека и животных физическими методами ус-
тановлено наличие собственного, биохимически пока не идентифицированного, фактора, который выполняет роль полимера, снижающего гидродинамическое сопротивление потока крови [238]. Длинные неветвящиеся молекулы такого рода полимеров встраиваются в поток любой жидкости, стабилизируя нестационарные и турбулентные потоки, уменьшая их вихреобразование, «застойные зоны» и, тем самым, снижают гидродинамическое сопротивление потока в соответствии с эффектом Томса (B. Toms, 1948). Внутривенное введение растворов таких полимеров животным позволяет уменьшить выраженность экспериментального атеросклероза, а также снизить постстенотические нарушения кровотока при коарктации аорты, увеличить сердечный выброс при неизменном или даже пониженном АД без увеличения ча- стоты сердечных сокращений, увеличить коллатеральное кровообращение в мозге [238]. Не исключено, что и в клинических условиях эти гемодинамические свойства потока крови также могут рассматриваться как характеристики тяжести течения нарушений мозгового кровообращения и атеросклеротического поражения МАГ в со- четании с артериальной гипертензией.
Оценка соотношения состояния всех МАГ с церебральной гемодинамикой позволила установить, что при суммарном сужении просвета артерий, превышающем 40%, наблюдается развитие более выраженной редукции мозгового кровотока в обоих полушариях мозга. При этом обратная корреляционная зависимость показателей церебрального кровотока от степени суммарного сужения МАГ более тесная и составляет – 0,52 мл/100 г/мин на 1% суммарного стеноза, причем эти данные относятся прежде всего к группе больных, у которых НМК развивалось по механизму хронической цереброваскулярной недостаточности [237].
Исследование церебральной гемодинамики в соче- тании с данными ангиографии и компьютерной томографии позволило оценить структурно-функциональную сущность гемодинамического паттерна* при нарастающем окклюзирующем процессе в ВСА и остальных МАГ. Основу гемодинамического паттерна составляют умеренная равномерная редукция мозгового кровотока в обоих полушариях головного мозга, отсутствие межполушарной и межрегионарной асимметрии кровотока, сохранность зависимости полушарного и регионарного кровотока гомологичных регионов бассейнов передней и средней мозговых артерий и зависимость церебральной гемодинамики от степени уменьшения суммарного просвета МАГ.
Патогенетическим «ядром» многообразных клини- ческих форм НМК является ишемия мозга. Она может быть диффузной или очаговой, преходящей или стойкой, однократной или повторной, мелкоили крупнооча- говой, обусловливающей неполный некроз ткани мозга или его инфаркт. Все эти изменения могут локализоваться в бассейне одной или нескольких мозговых артерий каротидной или вертебрально-базилярной системы. Сле-
4.2. Ишемическая болезнь головного мозга |
123 |
дует подчеркнуть, что указанные характеристики ишемии мозга не являются альтернативными. В практических условиях чаще встречаются их различные сочетания: инфаркт мозга, как правило, возникает на фоне предшествующих диффузных изменений мозга; эпизоды НМК часто бывают повторными; очаговые и диффузные изменения обнаруживаются в бассейнах нескольких артерий одной или обеих систем МАГ у одного и того же больного и т.п.
Пусковым механизмом повреждающего действия ишемии на мозговую ткань является снижение уровня высокоэнергетических фосфатов. Недостаток кислорода стимулирует переход на анаэробный гликолиз, обеспечивающий сохранение возможности синтеза АТФ и приводящий к накоплению молочной кислоты, что ведет к выраженному лактацидозу. Это проявляется в снижении величины рН в ткани мозга. Как известно, рН влияет на ауторегуляцию локального мозгового кровотока, в свою очередь, обеспечивает доставку кислорода к ткани. Кроме того, рН определяет функционирование клеточных мембран и активности ферментов, участвующих в гликолизе. Важно, что в условиях неполной ишемии снабжение мозга глюкозой сохраняется, этим поддерживается анаэробный гликолиз, ведущий к усилению лактацидоза и углублению поражения нейронов [207].
Âпроцессе развития ишемии избирательно нарушаются механизмы синаптической передачи. В мозге увеличивается внеклеточная концентрация гамма-амино- масляной кислоты и глутамата. В ишемизированной ткани уменьшается синтез дофамина и норадреналина, а высвобождение серотонина намного возрастает. Все это приводит к нарушению ауторегуляции мозгового кровотока, развитию вазоспазма, усилению агрегации тромбоцитов и формированию внутрисосудистого стаза, что углубляет ишемию, делает ее необратимой. Кроме того, высвобождение катехоламинов на пресинаптическом уровне может, вероятно, вызывать усиление активности нейронов и возникновение дополнительных потребностей в энергетических субстратах, что в условиях дефицита кровоснабжения усугубляет ишемическое поражение мозга [239].
Âпроцессе перекисного окисления липидов повышается образование простаноидов и лейкотриенов. В конечном итоге это ведет к изменениям сосудистой реактивности, нарушению проницаемости сосудов, повышению агрегационной способности тромбоцитов, микротромбозу и еще большему усугублению ишемии [206, 239, 240].
Таким образом, в процессе изменения клеточных структур при ишемии ключевое значение имеют: токси- ческое воздействие на клетку избыточного накопления возбуждающих аминокислот (эксайтотоксичность – от англ. to excite – возбуждать); лавинообразное поступление ионов кальция в клетку, распад клеточных мембран, накопление свободных радикалов, продуктов перекисного окисления липидов и эйкозаноидов [207, 241].
Изучение механизмов гибели нейронов при гипоксии и ишемии привлекло внимание к генетически запрограммированной клеточной смерти – апоптозу (от англ. apoptosis – утрата клеток). В экспериментальных исследованиях показано, что по механизму апоптоза могут погибать нервные клетки в зоне ишемии. Это связано с разобщением нервных клеток с другими клетками, в результате чего они лишаются необходимого нейротрофического фактора. Апоптоз, фрагментация ДНК и программированная клеточная смерть являются родственными, но не обязательно совпадающими процессами [242].
Преходящие нарушения мозгового кровообращения при поражении сонных артерий, или транзиторные ишемические атаки (ТИА), в 30–40-å годы расценивались как результат спазмов головных сосудов под влиянием патологических импульсов из области измененной ВСА.
Однако в дальнейшем стали высказываться серьезные сомнения в отношении роли ангиоспазмов в мозговой патологии. Указывалось, что мозговые артерии являются сосудами преимущественно эластического типа со слабо развитым мышечным слоем, и что они очень скудно снабжены нервными волокнами. В результате этих и некоторых других соображений многие невропатологи стали полностью отрицать патогенную роль церебральных ангиоспазмов.
С 50-х годов стало быстро завоевывать общее признание представление о сосудистой мозговой недостаточности и роли экстрацеребральных факторов. Согласно этому определению, преходящее НМК с очаговой симптоматикой возникает тогда, когда при наличии изменений сосудов мозга (чаще атеросклеротических), ведущих к дефициту мозгового кровотока, какой-либо экстрацеребральный стресс-фактор вызывает дополнительное его снижение. Это происходит обычно в результате падения системного АД, при ослаблении сердеч- ной деятельности, а также при перераспределении объема циркулирующей крови к периферическим частям тела (например, в горячей ванне). Перечисленные факторы, вызывая ограничение притока крови ко всему мозгу, ведут к его ишемии, особенно в зоне наиболее склерозированных сосудов, и к появлению очаговых неврологи- ческих симптомов [243]. Первоначально описанному механизму придавалось исключительное значение, однако на практике нередко встречались случаи, когда ТИА возникали не при падении, а при повышении системного АД. Подобные факты заставили вернуться к представлению о церебральных ангиоспазмах в патогенезе преходящих ишемических НМК.
Констрикция магистральных и других крупных артерий мозга типа спазма описана в многочисленных экспериментальных работах. Клинические наблюдения также показали, что при различных воздействиях (хирургические манипуляции на сосудах мозга, ангиография) спазм развивался легче в крупных, чем в пиальных
Рекомендовано к изучению сайтом МедУнивер - https://meduniver.com/

124 Глава 4. ПАТОГЕНЕЗ, ПАТОФИЗИОЛОГИЯ И КЛИНИКА КОРОНАРНОГО И КАРОТИДНОГО АТЕРОСКЛЕРОЗА
артериях. Дальнейшие исследования показали, что в условиях одновременного воздействия нескольких возмущающих факторов (гипоксия, повышение АД) различ- ные части артериальной системы мозга выполняют при этом лишь присущую им функцию. Пиальные артерии расширялись, регулируя адекватный приток крови в ткань мозга; магистральные артерии суживались, компенсируя, в свою очередь, повышение системного АД или избыточное кровенаполнение сосудов мозга. Артерии, расположенные в толще коры мозга, во всех случа- ях имели тенденцию к сужению или не меняли своего просвета [244].
Âобщем патофизическом аспекте под патологической реакцией мозговых артерий подразумевается такое функциональное поведение этих сосудов в виде ангиспазма или патологической дилатации, которое не обеспечивает регулирования кровотока, а ведет к его нарушениям. В случае недостаточного коллатерального притока крови патологическая вазоконстрикция может приводить к дефициту кровоснабжения соответствующих областей мозга.
Âпоследнее время все более склоняются к мысли о том, что патологический вазоспазм – это, скорее, патология эндотелия, выражающаяся в его длительном набухании, а не состояние продолжительного сокращения гладкомышечных волокон медии. Сосуды с поврежденным эндотелием сами обладают склонностью к спазмам
èменяют свою реактивность к ряду агентов [107, 223]. Вообще оказалось, что при ишемии и спазме наибо-
лее ранимой частью мозга являются не нервные клетки, а эндотелий капилляров, и причина умирания мозга – в микроциркуляторных нарушениях [223, 245]. Возникающее при спазме повреждение эндотелия ведет к нарушению синтеза простациклина, расширяющего сосуды. К другим гуморальным посредникам спазма относят тромбоксан, ангиотензин II, серотонин; наиболее выраженным спазмогенными свойствами обладают именно тромбоциты [246].
За последние годы особенно большое внимание уделяется третьему возможному механизму ТИА – микроэмболиям [245]. При этом речь идет не об эмболиях из сердца, о которых будет сказано ниже, а об артерио-ар- териальных эмболиях из атероматозных бляшек МАГ.
Материалом для микроэмболий служат кристаллы холестерина, атероматозные массы распадающейся бляшки, элементы пристеночного тромба. Недавними исследованиями показано, что такими микроэмболами чаще являются конгломераты тромбоцитов [207, 245, 246]. Оседая на измененном участке сосудистой стенки и соприкасаясь с ее коллагеновой субстанцией, изменяющиеся тромбоциты выделяют ряд сильных агрегантов, таких как тромбоксан и аденозиндифосфорная кислота. Под их воздействием происходит дальнейшая агрегация тромбоцитов с образованием крупных скоплений, становящихся материалом для эмболии. Остановка эмбола в мелком сосуде, сопровождаясь перифокальным отеком,
ведет к появлению очаговых неврологических симптомов, которые исчезают, когда эмбол распадается или лизируется, а отек мозговой ткани постепенно уменьшается или когда устанавливается полноценное коллатеральное кровообращение [243].
Âряде случаев эпизоды ТИА, наступающие без уловимых колебаний системного АД, обусловливаются изменениями физико-химических свойств крови: повышением ее вязкости, уменьшением содержания кислорода, гипогликемией. Эти факторы в условиях сниженного уровня кровоснабжения мозга могут приводить к уменьшению количества доставляемого к мозговой ткани кислорода и глюкозы ниже критического уровня, к задержке продуктов метаболизма, особенно в бассейне склерозированного сосуда, что ведет к появлению очаговых неврологических симптомов [206, 212, 243]. Эти механизмы могут иметь место при полицитемии, нарушении функции легких и некоторых других состояниях.
И все же наиболее часто ТИА наблюдаются при атеросклеротическом стенозирующем процессе в позвоноч- ных и сонных артериях.
Прогностическое значение ТИА зависит от нескольких факторов: от того, каким заболеванием они вызваны, какие механизмы лежат в их основе и, разумеется, от того, насколько эффективными были мероприятия, направленные на профилактику острого ишемического нарушения церебральной гемодинамики.
Ишемический инсульт – понятие, которое характеризует факт развития заболевания, обусловленного уменьшением кровотока в определенной зоне мозга, и приводящего к формированию ограниченного инфаркта мозговой ткани [247].
Инфаркт мозга – это зона некроза, образовавшаяся вследствие тяжелых нарушений метаболизма нейрональных и глиальных* структур, возникших в результате недостаточного кровообращения из-за стеноза (окклюзии) МАГ или артерий мозга, или из-за их тромбоза или эмболии, приводящих к дефициту перфузионного давления, резкому ограничению доставки оксигенированной крови, ишемии, гипоксии и гибели участка мозговой ткани.
Âсоответствии с современными представлениями о патогенетической гетерогенности ишемических инсультов [224], их развитие в 30–40% случаев связано с атеросклеротическим поражением МАГ и артерий мозга, причиной 15–20% инсультов являются кардиогенные эмболии, 25–30% случаев составляют лакунарные инфаркты как результат изменений внутримозговых сосудов при артериальной гипертонии, и 10% ишемических инсультов могут быть вызваны гемореологическими нарушениями, в частности коагулопатиями [248]. По некоторым зарубежным данным, тромбоз атеросклеротически измененных церебральных артерий является причиной 60% всех инфарктов мозга [249].
При уменьшении или полном прекращении кровотока в артерии развивается резко выраженная ишемия и
4.2. Ишемическая болезнь головного мозга |
125 |
тканевая гипоксия, возникают очаги полного ишемического некроза (инфаркта) мозга. Они могут локализоваться в любом отделе головного мозга. Их величина зависит от диаметра пораженного сосуда и возможностей коллатерального кровообращения. В пределах очага инфаркта происходит гибель всех структурных элементов нервной ткани – нейронов, волокон, нейроглии, капилляров, мелких артерий и вен. В участках мозга, прилегающих к зоне инфаркта, наблюдается неполный некроз, протяженность которого от границы некроза может колебаться в широких пределах (от нескольких микрон до нескольких миллиметров) [207].
Локализация инфарктов и очагов неполного некроза во многом зависит от особенностей строения артериальной системы мозга. Прежде всего следует подчеркнуть, что его кровоснабжение осуществляется в соответствии
ñангиогенетическим законом Шпальтегольца (W. Spalteholz, 1923). Согласно этому закону, сосуды всех полых органов, закладывающихся в эмбрионе в виде трубки, располагаются на поверхности органа в виде сети, от которой под прямым углом отходят ветви, направляющиеся в толщу органа (цит. по [207]).
Вторым фактором, определяющим локализацию инфарктов и очагов неполного некроза, является удаленность от основного источника питания той области мозга, в которой они расположены. При уменьшении кровотока в этом источнике в самых худших условиях оказывается область мозга, наиболее удаленная от источ- ника. Такая закономерность названа принципом «последнего луга» Шнайдера-Цюльха (M. Schnaider, K. Zulch) [250]. Эти авторы сравнили ситуацию уменьшения кровотока в артерии с ситуацией снижения подачи воды в ирригационной системе орашаемых лугов: при уменьшении потока в основном канале в наихудших условиях оказывается последний от источника воды луг.
Âбассейне каждой из основных артерий мозга наиболее удаленной зоной является область ее анастомозов
ñдругой артерией на поверхности и в глубине мозга. При этом, если на поверхности мозга анастомозами являются ветви артерий, то в глубоких отделах анастомозируют только сосуды микроциркуляторного русла, так как здесь отсутствуют артерио-артериальные анастомозы.
При ИБГМ область с редуцированным кровотоком, или зона «последнего луга» анатомически строго не фиксирована. Она может перемещаться в определенных пределах в зависимости от локализации и темпа формирования стеноза или облитерации артерий мозга, функционирования путей коллатерального кровообращения, состояния центральной гемодинамики. От этих же факторов зависит и величина зоны «последнего луга», в которой недостаточный кровоток приводит к изменению и гибели отдельных нейронов, возникновению очагов неполного некроза или инфарктов мозга.
Следует указать, что нарушения физико-химичес- ких свойств крови, присущие атеросклерозу вообще, при острых нарушениях мозгового кровообращения
приобретают особое значение. Увеличение агрегации тромбоцитов и эритроцитов, повышение вязкости крови, гиперпротромбинемия, гиперлипидемия, изменение белкового коэффициента в сторону нарастания уровня альбуминов значительно затрудняют капиллярный кровоток в зоне локальной ишемии мозга, приводя к возникновению патологического феномена «невосстановленного» кровотока [206]. При этом, даже если исчезает причина, вызвавшая локальную ишемию, может нарушаться нормальная жизнедеятельность нейронов и развиваться инфаркт мозга. Сказанное определяет важность динамической оценки системы гемостаза в остром периоде ишемического инсульта [251].
Изменения состояния микроциркуляции, уменьшение напряжения кислорода в капиллярной крови соче- таются со снижением газотранспортных свойств гемоглобина, а также с нарушением процесса диссоциации оксигемоглобина. Все это приводит к существенному затруднению газообмена между кровью и тканью мозга с первых часов острой ишемии.
Одним из важнейших феноменов, изучаемых в настоящее время, является так называемая ишемическая «полутень» [252]. Сущность этого феномена состоит в следующем. С помощью радионуклидных методов исследования мозгового кровотока было установлено, что вокруг инфаркта существует зона, в пределах которой нейроны сохранены, но не функционируют, так как объемный кровоток в этой зоне достаточен для поддержания жизнеспособности нейронов, но недостаточен для их функционирования [253]. Эта зона названа ишеми- ческой полутенью или пенумброй (лат. penumbra – полутень вокруг крупного солнечного пятна – umbra). В области пенумбры у человека объемный мозговой кровоток снижен до 23–10 мл на 100 г вещества мозга в минуту при норме 58 мл/100 г/мин (табл. 11). Установлено также, что уменьшение кровотока ниже 15 мл/100 г/мин приводит к исчезновению электроэнцефалографи- ческих сигналов, а ниже 10 мл/100 г/мин – к инфаркту мозга [209, 254].
Чрезвычайно важно учитывать зависимость нарушений клеточного метаболизма от уровня кровотока в зоне ишемии. Такие взаимоотношения могут быть различными по выраженности и длительности.
Таблица 11
Пороги нарушения церебральной гемодинамики при ишемии головного мозга
Показатель |
Норма |
Порог |
Пен% |
Порог |
Инфаркт |
|
|
ишемии |
умбра |
инфаркта |
мозга |
|
|
|
|
|
|
Мозговой |
|
|
|
|
|
кровоток, |
50–60 |
2 0 |
15–10 |
10–8 |
0 |
мл/100 г/мин |
|
|
|
|
|
|
|
|
|
|
|
РО 2 в ткани |
|
|
|
|
|
мозга, |
35–45 |
4 0 |
35–30 |
30–25 |
0 |
мм рт.ст. |
|
|
|
|
|
|
|
|
|
|
|
Рекомендовано к изучению сайтом МедУнивер - https://meduniver.com/
126 Глава 4. ПАТОГЕНЕЗ, ПАТОФИЗИОЛОГИЯ И КЛИНИКА КОРОНАРНОГО И КАРОТИДНОГО АТЕРОСКЛЕРОЗА
Как установлено многочисленными исследованиями, ишемия, обусловленная снижением мозгового кровотока любого генеза, приводит к острому дефициту макроэргических соединений, таких как АТФ и фосфокреатин; угнетается аэробный путь утилизации глюкозы и активируется анаэробный путь энергообеспечения. В этих условиях основное количество глюкозы трансформируется в молочную кислоту, которая, накапливаясь, приводит к прогрессирующему лактоацидозу. Последний вызывает ряд патологических эффектов, в частности вазодилатацию и гиперперфузию в зоне ишемии, что в еще большей мере усугубляет нарушения метаболизма, а сочетание лактоацидоза с тканевой гипоксией дезорганизует функцию ферментной системы, управляющей ионным транспортом. На этом фоне развиваются грубые изменения дыхательной функции митохондрий и угасание электрического потенциала клеточных мембран. Этот патологический процесс влечет за собой пассивный отток ионов калия из нейронов и интенсивный приток в них ионов кальция, натрия и хлора, а также внутриклеточное накопление свободных жирных кислот
Ишемия
Гиперпродукция катехоламинов  Гипоксия
Гипоксия
Выключение дыхательной цепи митохондрий
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Активация |
|
|
|
|
|
|
|
|
|
|
Торможение окисления |
|
|||||||
гликолиза |
|
|
|
|
|
|
|
|
|
|
|
|
жирных кислот |
|
|||||
Накопление |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||
|
|
|
Дефицит АТФ |
|
|
|
|
|
|
|
|
|
|
|
|
||||
лактата |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||
|
|
|
|
|
|
|
|
|
|
Накопление |
|
||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
жирных кислот |
|
|||
|
|
|
|
|
|
Ингибирование |
|
|
|
|
|
|
|
|
|
|
|
|
|
Ацидоз |
|
|
|
гликолиза |
|
|
|
|
|
|
|
Появление |
|
||||||
|
|
|
|
|
|
|
|
|
|
|
|||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
мембранных |
|
|||
Нарушение |
|
|
|
Повышение |
|
|
|
|
|
|
|
и митохондриальных |
|
||||||
|
|
|
|
|
|
|
|
|
|
ферментов |
|
||||||||
ионного |
|
|
|
проницаемости |
|
|
|
|
|
|
|
|
|||||||
равновесия |
|
|
|
клеточных |
|
|
|
|
|
Выход плазматических |
|
||||||||
|
|
|
|
|
|
|
|
|
|||||||||||
в нейронах |
|
|
|
мембран |
|
|
|
|
|
|
|
ферментов |
|
||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Повреждающее |
|
|
|
|||
Накопление |
|
|
|
Активация |
|
|
|
|
|
|
|
действие |
|
|
|
||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||
ионов |
|
|
|
липаз |
|
|
|
|
|
|
|
жирных |
|
|
|
||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||
кальция |
|
|
|
|
|
|
|
|
|
|
|
|
кислот |
|
|
|
|||
в нейроне |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||
|
|
|
|
|
|
Повреждение |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
клеточных |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Усиление выхода |
|
|||||
|
|
|
|
|
|
мембран |
|
|
|
|
|
|
|
|
|||||
|
|
|
|
|
|
|
|
|
|
|
|
|
мембранных энзимов |
|
|||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Возрастание |
|
|||
|
|
|
|
|
|
Повреждение |
|
|
|
|
|
|
|
цитоплазматических |
|
||||
|
|
|
|
|
|
структур |
|
|
|
|
|
|
|
|
и |
|
|||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||
|
|
|
|
|
|
клеток |
|
|
|
|
|
|
|
митохондриальных |
|
||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
энзимов |
|
|||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||
|
|
|
|
Необратимые изменения |
|
|
|
|
|
|
|
||||||||
|
|
|
|
|
|
|
|
|
|
||||||||||
|
|
|
|
|
нейронов - некроз |
|
|
|
|
|
|
|
|||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
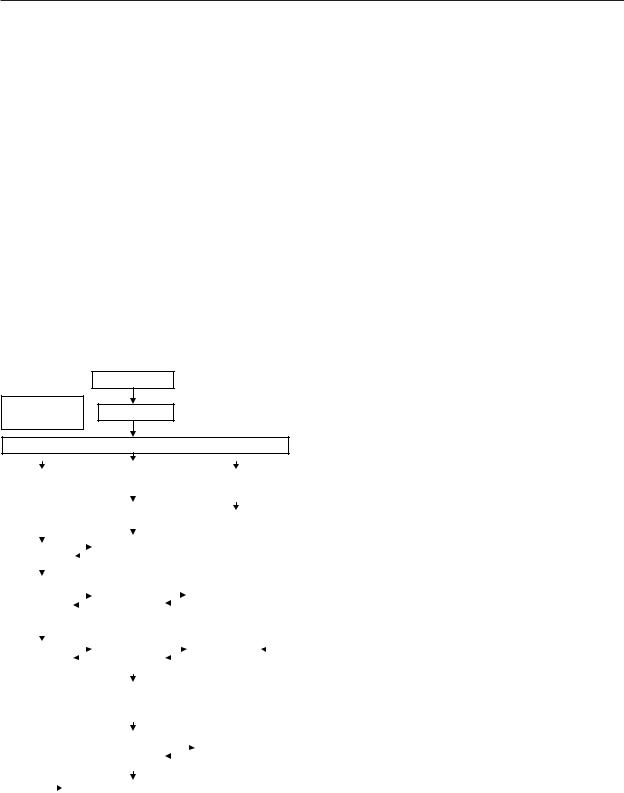
Рис. 64. Упрощенная схема нарушений метаболизма моз га при ишемическом инсульте [247]
и воды. Все это ведет к набуханию дендритов и лизису клеточных элементов нейронов – белковых и липидных комплексов аминокислот [255]. Повреждение мембранных структур интенсифицирует перекисное окисление липидов и накопление жирных кислот – процессы, от интенсивности которых в значительной степени зависят темпы и распространенность метаболических сдвигов, приводящих к формированию инфаркта (рис. 64).
Деполяризация клеточных мембран вызывает, кроме указанных изменений метаболизма, высвобождение возбуждающих нейротрансмиттеров – глютамата и аспартата. Они оказывают дополнительный цитотоксический эффект, усиливая приток ионов натрия и хлора в нейроны и, тем самым, способствуя их набуханию и функциональной дезорганизации. В острой стадии инсульта нарушается ферментативная активность организма; одним из ее проявлений является образование в нейронах свободных радикалов, повреждающих эндотелий сосудов [247].
В зависимости от степени ишемии активируется цикл арахидоновой кислоты с вторичным накоплением ее продуктов – простагландинов, лейкотриенов, тромбоксана. Последний, как известно, оказывает негативное влияние на сосудистый тонус и способствует активной агрегации тромбоцитов [222, 245, 256].
Помимо грубых локальных изменений непосредственно в зоне церебральной ишемии, патологические процессы распространяются на систему гуморальной и гормональной регуляции. Наиболее значима реактивная гипергликемия, практически не купируемая соответствующими препаратами [247]. Эти изменения способствуют гликозилированию гемоглобина, которое влечет за собой снижение газотранспортных свойств крови, что при дефиците в нейронах глюкозы и кислорода углубляет генерализованную дезорганизацию клеточного метаболизма.
Выраженная церебральная ишемия может сопровождаться повышением уровня катехоламинов в плазме крови. Однако в условиях резкой пассивной вазодилатации (вплоть до вазопаралича), обусловленной глубоким лактоацидозом, вазоконстрикторный эффект катехоламинов может проявляться в минимальной степени или вовсе отсутствовать.
Морфологические проявления ишемического поражения мозга в виде патологии отдельных нервных клеток, возникновения зон неполного некроза и инфарктов мозга, глиальной и макрофагальной реакций, сопровождающих эти процессы и их исходы, имеют универсальный характер и не зависят от их локализации в мозге или от источников кровоснабжения, будь то каротидная или вертебрально-базилярная артериальная система.
Атеросклеротическая энцефалопатия в системе ВСА характеризуется широким спектром морфологических изменений мозга, основными из которых являются: ганглиозноклеточные выпадения, очаги неполного некроза, малые инфаркты, включая малые поверхностные (гра-
4.2. Ишемическая болезнь головного мозга |
127 |
нулярная атрофия коры) и малые глубинные (лакунарные) инфаркты, средние, большие и обширные корковоподкорковые инфаркты мозга. Перечисленные формы церебральной патологии могут встречаться изолированно или, что бывает чаще, в различных сочетаниях одни с другими, а также с проявлениями ишемического поражения в бассейнах артерий вертебрально-базилярной системы и с различными формами гипертонической ангиоэнцефалопатии.
Момент возникновения инфаркта мозга, как правило, совпадает с временем появления выраженной и стойкой неврологической симптоматики. Следует отметить, что появление первых симптомов может опережать появление самого инфаркта и быть связанным с началом локальной ишемии, когда объемный кровоток снижается до 35–40 мл/100 г/мин (при норме, напоминаем, 58мл/ 100г/мин) [257].
Большинством исследователей предлагается выделять 3 стадии изменений ткани и формирования инфаркта мозга [207, 247, 257]:
•Первая стадия – развитие некроза; длится 2–3 суток
èхарактеризуется сморщиванием цитоплазмы* и кариоплазмы* клеток, а также различной степенью перифокального отека. Регресс неврологического дефекта в этой стадии, как правило, минимален.
•Вторая стадия – разжижение или резорбция некротических масс; длится несколько недель или месяцев, когда наблюдается паннекроз всех клеточных элементов, перифокальная пролиферация астроглии
èмелких сосудов. Возможности регрессии неврологического дефекта в этой стадии определяются преимущественно уровнем перифокальной васкуляризации, то есть активностью и развитием системы коллатерального кровообращения.
•Третья стадия – организация инфаркта, формирование рубца с полостью или без нее. Эта стадия не имеет определенной временной характеристики; по мере увеличения срока с момента заболевания на месте ишемического очага развиваются или глиальные рубцы, или кистозные дефекты ткани. Перспектива восстановления нервных функций зависит от состояния коллатерального кровообращения и возможности мобилизации функциональных резервов перифокальных зон мозга.
В заключение, хотя бы коротко, необходимо упомянуть о другой существующей форме острого нарушения кровообращения головного мозга. Геморрагический инсульт – кровоизлияние в мозг и/или подоболочечное (субарахноидальное) пространство встречается в 4–5 раз реже, чем ишемический инсульт, а основными его этиологическими факторами, наряду с артериальной гипертонией, могут стать врожденные и приобретенные артериальные и артериовенозные аневризмы. Субдуральные и эпидуральные гематомы чаще имеют травматический генез. Преимущественная локализация гематом – боль-
шие полушария головного мозга (около 90% паренхиматозных кровоизлияний), у 10% больных поражается ствол головного мозга или мозжечок. В большинстве случаев отмечается разрыв сосуда, значительно реже геморрагии происходят по типу диапедезных* кровоизлияний [207, 225, 247].
Атеросклероз ВСА приводит не только к острым ишемическим поражениям мозга, протекающим с клинической картиной инсульта, но и является причиной постепенного нарастания неврологических и психических нарушений вследствие длительной и стойкой НМК, нередко протекающей с повторными эпизодами острой ишемии мозга. Эта форма сосудистой патологии мозга
âотечественной литературе обозначается как дисциркуляторная энцефалопатия [243].
Дисциркуляторная энцефалопатия, развивающаяся на основе атеросклеротического поражения МАГ и артерий мозга, клинически проявляется в виде нарастающих интеллектуально-мнестических* расстройств вплоть до деменции* на фоне разнообразных неврологических симптомов, обусловленных предшествующими очаговыми и диффузными изменениями мозга ишемического и гипоксического характера.
Âткани мозга при этом обнаруживаются участки с ишемически измененными нервными клетками или уча- стки выпадения нейронов с развитием глиоза. Встреча- ются мелкие очажки некроза, иногда с замещением нервной ткани глиозными рубчиками. В результате гибели нервной ткани, преимущественно вокруг мелких сосудов, образуются периваскулярные небольшие полости (лакуны). При наличии большого количества таких лакун, что часто наблюдается в областях подкорковых узлов, нервная ткань может принять губчатый вид («status lacunaris»). Может развиваться и гранулярная атрофия коры [243].
Основной причиной развития деменции являются множественные инфаркты, локализующиеся в коре мозга и в подкорковых структурах. Инфаркты, приводящие к деменции, могут быть малыми поверхностными, множественными, малыми глубинными, а также средними и большими.
Наряду с деменцией дисциркуляторная энцефалопатия может проявляться также в виде псевдобульбарного паралича, который развивается при множественных двусторонних инфарктах, локализующихся в области кортикоядерных волокон. При этом, кроме инфарктов различной величины, давности и локализации, обнаруживаются косвенные признаки уменьшения массы мозга – расширение желудочков и борозд полушарий головного мозга, что, однако, наблюдается не всегда, особенно при единичных мелких инфарктах [207].
Некоторые инфаркты при атеросклеротической дисциркуляторной энцефалопатии протекают бессимптомно. Эти «немые» инфаркты локализуются, как правило,
âглубоких отделах мозга. Большая их часть является малыми глубинными (лакунарными) инфарктами, мень-
Рекомендовано к изучению сайтом МедУнивер - https://meduniver.com/

128 Глава 4. ПАТОГЕНЕЗ, ПАТОФИЗИОЛОГИЯ И КЛИНИКА КОРОНАРНОГО И КАРОТИДНОГО АТЕРОСКЛЕРОЗА
шая часть относится к средним корково-подкорковым инфарктам. «Немые» инфаркты встречаются у 34,5% больных с выраженным атеросклерозом ВСА, формируются медленно, по-видимому, во время сна или в период бодрствования при минимальной активности больного [258]. «Немые» инфаркты встречаются у 11% больных, перенесших инсульт; они играют определенную роль в развитии сосудистой мультиинфарктной деменции, и способствует также развитию тяжелых НМК
[259].«Немые» инфаркты, как и инфаркты, сопровождающиеся клинической симптоматикой, гетерогенны, имеют различную локализацию; в процессе своей организации они эволюционируют так же, как и «симптомные» инфаркты мозга.
Артериальная гипертония, как известно, приводит не только к острым геморрагическим НМК, но и к постепенному нарастанию неврологических и психических расстройств вследствие хронической недостаточности кровоснабжения мозга. Для обозначения такой патологии в отечественной литературе используются термины «гипертоническая дисциркуляторная энцефалопатия» [225], «хроническая гипертоническая энцефалопатия»
[260].За рубежом для обозначения хронической недостаточности кровоснабжения мозга при артериальной гипертонии чаще используются различные термины, которые отражают одно из наиболее ярких клинических проявлений этой патологии – деменцию: мультиинфарктная деменция, лакунарная деменция, хроническая прогрессирующая субкортикальная энцефалопатия Бинсвангера и др. Под дисциркуляторной энцефалопатией обыч- но понимают прогрессирующую диффузную и мелко- очаговую патологию мозга, обусловленную нарастающим ухудшением его кровоснабжения.
В связи с большой распространенностью артериальной гипертонии в популяции, тенденцией к старению населения и с увеличением числа хронических форм цереброваскулярной патологии, удельный вес гипертонической дисциркуляторной энцефалопатии в общей структуре НМК остается высоким [207].
В случаях, когда артериальная гипертония развивается в 20–25-летнем возрасте, начальные симптомы гипертонической энцефалопатии могут регистрироваться уже к 30 годам. В старших возрастных группах они достигают степени сосудистой деменции, поражая, по данным разных авторов, 1,5–16,3% популяции, и этот процент быстро возрастает после 70 лет [225].
По мере формирования гипертонической дисциркуляторной энцефалопатии распространенность и выраженность очагов дегенеративных изменений в глубоких отделах белого вещества обоих полушарий резко возрастает, достигая наибольшей тяжести у больных деменцией в виде феномена лейкоареоза (греч leukos – белый, araios – редкий, неплотный). Считается, что в основе лейкоареоза лежат повышение содержания воды в белом веществе, его вакуолизация и развитие микроскопических полостей, заполненных жидкостью (спонги-
оз). Дальнейшее прогрессирование выраженности лейкоареоза связано с замещением гибнущих волокон белого вещества мозга пролиферирующими астроцитами*, содержащими большое количество воды, и макрофагами, содержащими липиды (зернистые шары) [207]. Однако, по данным компьютерной и магнито-резонансной томографии мозга, лейкоареоз довольно часто выявляется у лиц пожилого возраста, в том числе при отсутствии нарушения психических функций. Это, конечно, затрудняет клиническую интерпретацию лейкоареоза, как нейровизуализационного феномена.
Диффузная патология белого вещества полушарий головного мозга занимает ведущее место в развитии нарушений психических функций у больных с хронической цереброваскулярной патологией при артериальной гипертонии, являясь, по существу, морфологической основой сосудистой деменции подкоркового типа. Выделены две основные формы подкорковой сосудистой деменции, протекающие с преимущественной патологией белого вещества:
1)мультиинфарктная деменция, для которой, как уже было сказано, характерны множественные глубинные (лакунарные) инфаркты;
2)субкортикальная артериосклеротическая энцефалопатия (болезнь Бинсвангера), в основе которой лежат диффузные изменения белого вещества, сопровождающиеся утратой миелина и уменьшением объема белого вещества.
Деменция при болезни Бинсвангера относится к подкорковому типу, обусловленному нарушением связи в целом сохранной коры мозга с нижерасположенными активирующими структурами, в первую очередь с таламусом. В клинической практике около 70% всех случа- ев деменции, возникающих в пожилом возрасте, связаны либо с болезнью Бинсвангера, как одной из форм сосудистой мозговой патологии, либо с болезнью Альцгеймера, относящимся к первичным дегенеративным формам деменции, либо с сочетаниями сосудистых мозговых изменений и нарушений, характерных для болезни Альцгеймера. Диагностика последних особенно трудна.
Значительные изменения в коре мозга с массивной гибелью нейронов при болезни Альцгеймера приводят к неуклонно прогрессирующему нарушению памяти и необратимому нарушению психических функций. При болезни Бинсвангера кора полушарий мозга остается в целом сохранной, и развитие деменции обусловлено разобщением различных отделов коры и подкорковых структур вследствие первичного диффузного поражения белого вещества полушарий. При улучшении кровоснабжения белого вещества можно предупредить дальнейшее прогрессирование болезни Бинсвангера [262]. В связи с этим, в отличие от первичной дегенеративной деменции типа Альцгеймера, деменция сосудистого типа Бинсвангера в настоящее время рассматривается как
4.2. Ишемическая болезнь головного мозга |
129 |
форма патологии, потенциально поддающаяся коррекции.
Классификация и клинические проявления сосудистых поражений головного мозга
Клинические проявления мозговой сосудистой недостаточности, обусловленной атеросклеротическим поражением МАГ и артерий мозга, очень разнообразны. Они охватывают многочисленные острые и хронически протекающие, прогрессирующие формы ишемических НМК: начальные проявления недостаточности мозгового кровообращения, преходящие НМК, малый инсульт, мозговой инсульт с необратимым неврологическим дефицитом различной степени тяжести, повторные ТИА и инсульты, асимптомные (немые) инфаркты мозга, а также медленно прогрессирующая недостаточность кровоснабжения мозга – дисциркуляторная энцефалопатия.
Использование общепринятой классификации сосудистых поражений головного мозга диктуется необходимостью единообразия в формулировках диагнозов, позволяющего правильно осуществлять статистическую обработку и анализ медицинских документов, сопоставлять результаты деятельности различных лечебных уч- реждений, проводить клинико-эпидемиологические исследования.
В настоящее время нормативным документом является изданная в 1995 г. «Международная статистическая классификация болезней и проблем, связанных со здоровьем» Х пересмотра [204], в которой сосудистые поражения головного мозга разнесены по двум рубрикам.
Класс IX болезни системы кровообращения (100–199) Цереброваскулярные болезни (160 – 169)
160Субарахноидальное кровоизлияние
161Внутримозговое кровоизлияние
162Другое нетравматическое внутричерепное кровоизлияние
163Инфаркт мозга
164Инсульт, не уточненный как кровоизлияние или инфаркт
165Закупорка и стеноз прецеребральных артерий, не приводящие к инфаркту мозга
166Закупорка и стеноз церебральных артерий, не приводящие к инфаркту мозга
168Поражения сосудов мозга, классифицированных в других рубриках
169Последствия цереброваскулярной болезни
Класс VI болезни нервной системы (G 00 – G 99)
G 45 Преходящие транзиторные церебральные ишемические приступы (атаки) и родственные синдромы
G 45.0 Синдром вертебробазилярной артериальной системы
G 45.1 Синдром сонной артерии (полушарный)
G 45.2 Множественные и двусторонние синдромы церебральных артерий
G 45.3 Преходящая слепота
G 45.4 Транзиторная глобальная амнезия
G 45. Другие транзиторные церебральные ишеми- ческие атаки и связанные с ними синдромы G 45.9 Транзиторная церебральная ишемическая
атака неуточненная
G 46 Сосудистые мозговые синдромы при цереброваскулярных болезнях
G 46.0 Синдром средней мозговой артерии G 46.1 Синдром передней мозговой артерии G 46.2 Синдром задней мозговой артерии
G 46.3 Синдром инсульта в стволе головного мозга G 46.4 – G 46.7
G 46.8 Другие сосудистые синдромы головного мозга при цереброваскулярных болезнях.
В нашей стране широкое распространение получила классификация сосудистых поражений головного и спинного мозга, предложенная в 1985 г. Е.В. Шмидтом [263]; в ней учтены формы патологии, не учитываемые МКБ.
À.Начальные проявления недостаточности кровоснабжения мозга
Á.Преходящие нарушения мозгового кровообращения
1.Транзиторные ишемические атаки
2.Гипертонические церебральные кризы
2.0.Общемозговые
2.1.С очаговыми нарушениями
Â.Инсульт
1.Субарахноидальное нетравматическое кровоизлияние
2.Геморрагический инсульт
2.0.Кровоизлияние в головной мозг
3.Другие и неуточненные внутричерепные кровоизлияния
4.Ишемический инсульт (инфаркт)
4.0.Церебральный ишемический инсульт
4.0.0.При поражении МАГ
4.0.1.При поражении церебральных артерий
4.0.2.При эмболии церебральных артерий
5.Инсульт с восстановимым неврологическим дефицитом (малый инсульт)
Ã.Прогрессирующие нарушения мозгового кровообращения
1.Хроническая субдуральная гематома
2.Дисциркуляторная энцефалопатия
2.0.Атеросклеротическая
2.1.Гипертоническая
2.2.Венозная и другие или неуточненные
Д. Другие и неуточненные сосудистые поражения
Надо заметить, что и в наши дни вопросы классификации в клинической ангионеврологии широко дискутируются в периодической научной печати [264, 265], и, учитывая внедрение в клинику все новых методов прижизненной визуальной диагностики морфологического
Рекомендовано к изучению сайтом МедУнивер - https://meduniver.com/