

Книги по МРТ КТ на английском языке / PLUM AND POSNER S DIAGNOSIS OF STUPOR AND COM-1
.pdf46 Plum and Posner’s Diagnosis of Stupor and Coma
medulla.28 In addition, the nucleus of the solitary tract provides both direct and relayed excitatory inputs to the cardiac decelerator neurons in the nucleus ambiguus.27 Thus, a rise in blood pressure results in a reflex fall in heart rate and vasomotor tone, re-establishing a normal arterial pressure. Conversely, a fall in blood pressure causes a reflex tachycardia and vasoconstriction, re-establishing the necessary arterial perfusion pressure. As a result, on assuming an upright posture, there is normally a small increase in both heart rate and blood pressure.
On occasion, loss of consciousness may result from failure of this baroreceptor reflex arc. In such patients, measurement of standing and supine blood pressure and heart rate discloses a fall in blood pressure on assuming an upright posture that is clinically associated with symptoms of insufficient CBF. Rigid criteria for diagnosing orthostatic hypotension (e.g., a fall in blood pressure of 10 or 15 mm Hg) are not useful, as systemic arterial pressure is usually measured in the arm but the symptoms are produced by decreased blood flow to the brain. A pressure head that is adequate to perfuse the arm (which is at the same elevation as the heart) will be reduced by 15 to 23 mm Hg at the brain in an upright posture, and if perfusion pressure to the brain falls even a few mm Hg below the level needed to maintain autoregulation, the drop in cerebral perfusion may be precipitous.
The most common nonneurologic causes of orthostatic hypotension, including low intravascular volume (often a consequence of diuretic administration or inadequate fluid intake), cardiac pump failure, and medications that impair arterial constriction (e.g., alpha blockers or direct vasodilators), do not impair the tachycardic response. Most neurologic cases of orthostatic hypotension, including peripheral autonomic neuropathy or central or peripheral autonomic degeneration, impair both the heart rate and the blood pressure responses. Put in other words, the hallmark of baroreceptor reflex failure is absence of the elevation of heart rate when arterial pressure falls in response to an orthostatic challenge.
Respiration
The brain cannot long survive without an adequate supply of oxygen. Within seconds of
being deprived of oxygen, brain function begins to fail, and within minutes neurons begin to die. The physician must ensure that respiration is supplying adequate oxygenation. To do this requires examination of both respiratory exchange and respiratory pattern. Listening to the chest will ensure that there is adequate movement of air. A normal patient at rest will regularly breathe at about 14 breaths per minute and the exchange of air can be heard at both lung bases. The physician should estimate from the rate and depth of respiration whether the patient is hypoor hyperventilating or whether respiration is normal. The patient’s color is a gross indicator of oxygenation: cyanosis indicates deficient oxygenation; a cherry red color may also indicate deficient oxygenation because of CO intoxication. A better estimate of oxygenation can be achieved by placing an oximeter on the finger; many intensive care units and some emergency departments also measure expired CO2, which correlates well with PCO2.
This section considers the neuroanatomic basis of respiratory abnormalities that accom-
Table 2–2 Neuropathologic Correlates
of Breathing Abnormalities
Forebrain damage
Epileptic respiratory inhibition
Apraxia for deep breathing or breath holding ‘‘Pseudobulbar’’ laughing or crying Posthyperventilation apnea
Cheyne-Stokes respiration Hypothalamic-midbrain damage
Central reflex hyperpnea (neurogenic pulmonary edema)
Basis pontis damage
Pseudobulbar paralysis of voluntary control Lower pontine tegmentum damage or
dysfunction Apneustic breathing Cluster breathing
Short-cycle anoxic-hypercapnic periodic respiration
Ataxic breathing (Biot) Medullary dysfunction
Ataxic breathing
Slow regular breathing
Loss of autonomic breathing with preserved voluntary control
Gasping

Examination of the Comatose Patient |
47 |
pany coma (Table 2–2, Figure 2–3). Chapter 5 discusses respiratory responses to metabolic disturbances. Because neurogenic and metabolic influences on breathing interact extensively, respiratory changes must be interpreted cautiously if there is evidence of pulmonary disease.
The pattern of respiration can give important clues concerning the level of brain damage. Once assured that there is adequate exchange of oxygen, the physician should watch
the patient spontaneously breathe. Irregularities of the respiratory pattern that provide clues to the level of brain damage are described in the paragraphs below.
PATHOPHYSIOLOGY
Breathing is a sensorimotor act that integrates nervous influences arising from nearly every level of the brain and upper spinal cord. In humans, respiration subserves two major func-
Cortex
Infralimbic
Cortex
VP Thalamus
Insular
Cortex
Hypothalamus |
Amygdala |
|
|
|
|
Midbrain |
Parabrachial Nucleus |
|
|
|
|
Pons |
Nucleus Ambiguus |
IX and X Nerves |
Rostral Ventrolateral |
Xn. |
|
Medulla |
|
|
|
|
|
Medulla |
|
|
Nucleus of the |
|
|
Solitary Tract |
|
ACh |
|
Caudal |
Muscarinic |
|
|
|
Spinal Cord: |
Ventrolateral |
|
Cervical |
Medulla |
|
|
|
|
|
|
1-receptor |
Thoracic |
|
|
Lumbar |
NE |
1-receptor |
|
||
Sacral
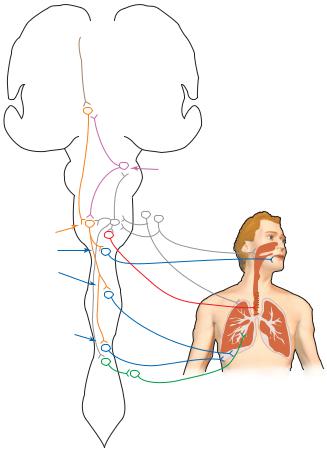
Figure 2–3. A diagram summarizing the cardiovascular control pathways in the brain. Visceral afferent information (gray) arrives from nerves IX and X into the nucleus of the solitary tract. This information is then distributed to the parabrachial nucleus, which relays it to the forebrain, and to the ventrolateral medulla, where it controls cardiovascular reflexes. These include both vagal control of heart rate (red) and medullary control (purple) of the sympathetic vasomotor control area of the rostral ventrolateral medulla (orange), which regulates sympathetic outflow to both the heart and the blood vessels (dark green). Forebrain areas that influence the cardiovascular system (brown) include the insular cortex (a visceral sensory area), the infralimbic cortex (a visceral motor area), and the amygdala, which produces autonomic emotional responses. All of these act on the hypothalamic sympathetic activating neurons (light green) in the paraventricular and lateral hypothalamic areas to provide behavioral and emotional influence over the blood pressure and heart rate. ACh, acetylcholine; NE, norepinephrine; VP, ventroposterior.
48 Plum and Posner’s Diagnosis of Stupor and Coma
tions: one of metabolism and the other behavioral. Metabolically, respiratory control is directed principally at maintaining tissue oxygenation and normal acid-base balance. It is regulated mainly by reflex neural mechanisms located in the posterior-dorsal region of the pons and in the medulla. Behavioral control of breathing allows it to be integrated with swallowing, and in humans, with verbal and
emotional communication as well as other behaviors.
Respiratory rhythm is an intrinsic property of the brainstem that is generated by a network of neurons that lie in the ventrolateral medulla, including the pre-Bo¨tzinger complex29,30 (see Figure 2–3). This rhythm is regulated in the intact brain by a number of influences that enter via the vagusand glossopharyngeal nerves.
Cortex
 Prefrontal
Prefrontal
Cortex
Hypothalamus
Midbrain |
Parabrachial Nucleus |
|
Pons |
||
|
||
Spinal Cord: |
ACh |
|
Cervical |
ACh Muscarinic |
|
T2-T12: Intercostal Motor Neurons |
Nicotinic |
|
Thoracic |
|
|
T2-8 |
|
|
Lumbar |
NE |
|
Medulla |
IX and X Nerves |
|
Ventral Respiratory Group |
Nucleus |
|
Hypoglossal Motor Nucleus |
Ambiguus |
|
C3-5: Phrenic Motor Nucleus |
|
|
|
2 adrenergic |
Sacral
Figure 2–4. A diagram summarizing the respiratory control pathways in the brain. Afferents from the lung (pulmonary stretch), upper airway (cough reflexes), and carotid body arrive via cranial nerves IX and X in the nucleus of the solitary tract (gray), also called the dorsal respiratory group. These control airway and respiratory reflexes, analogous to the cardiovascular system, by inputs to the ventrolateral medulla. These include outputs to the airways via the vagus nerve (red) and outputs from the ventral respiratory group (orange) to the spinal cord, controlling sympathetic airway responses (green) and respiratory motor (phrenic motor nucleus, blue) and accessory motor (hypoglossal and intercostal, blue) outputs. The ventral respiratory group is responsible for generating respiratory rhythm. However, it is assisted in this process by the parabrachial nucleus (or pontine respiratory group, purple), which receives ascending respiratory afferents and integrates them with other brainstem reflexes (e.g., swallowing). The prefrontal cortex (brown) provides behavioral regulation of breathing, producing a continual breathing rhythm even in the absence of metabolic need. This influences the hypothalamus (light green), which may vary respiratory pattern in coordination with behavior or emotion. ACh, acetylcholine; NE, norepinephrine.
The carotid sinus branch of the glossopharyngeal nerve brings afferents that carry information about blood oxygen and carbon dioxide content, whereas the vagus nerve conveys pulmonary stretch afferents. These terminate in the commissural, ventrolateral, intermediate, and interstitial components of the nucleus of the solitary tract.31–33 Chemoreceptor afferents can increase respiratory rate and depth, whereas pulmonary stretch receptors tend to inhibit lung inflation (the Herring-Breuer reflex). These influences are relayed to reticular areas in the ventrolateral medulla that regulate the onset of inspiration and expiration.34 In addition, serotoninergic neurons in the ventral medulla may also serve as chemoreceptors and directly influence the nearby circuitry that generates the respiratory rhythm.35,36
The medullary circuitry that controls respiration is under the control of pontine cell groups that integrate breathing with ongoing orofacial stimuli and behaviors.37 Neurons in the parabrachial nucleus primarily increase the rate and depth of respiration, presumably in relation to emotional responses or in anticipation of metabolic demand during various behaviors. On the other hand, neurons located more ventrally in the intertrigeminal zone, between the principal sensory and motor trigeminal nuclei, produce apneas, which are necessary during swallowing and in response to noxious chemical irritation of the airway (e.g., smoke or water in the nasal passages).38
Superimposed upon these metabolic demandsand basic reflexes,theforebraincancommand a wide range of respiratory responses. Respiration can be altered by emotional response, and it increases in anticipation of metabolic demand during voluntary exercise, even if the muscle that is to be contracted has been paralyzed (i.e., as a consequence of central command rather than metabolic reflex). The pathways that control vocalization in humans appear to originate in the frontal opercular cortex, which provides premotor and motor integration of orofacial motor actions. However, there is also a prefrontal contribution to the maintenance of respiratory rhythm, even in the absence of metabolic demand (the basis for posthyperventilation apnea, described below).
These considerations make the recognition of respiratory changes useful in the diagnosis of coma (Figure 2–5).
Examination of the Comatose Patient |
49 |
POSTHYPERVENTILATION APNEA
If the arterial carbon dioxide tension is lowered by a brief period of hyperventilation, a healthy awake subject will nevertheless continue to breathe with a normal rhythm, at least initially,39 albeit at reduced volume, until the PCO2 returns to its original level. By contrast, subjects with diffuse metabolic impairment of the forebrain, or bilateral structural damage to the frontal lobes, commonly demonstrate posthyperventilation apnea.40 Their respirations stop after deep breathing has lowered the carbon dioxide content of the blood below its usual resting level. Rhythmic breathing returns when endogenous carbon dioxide production raises the arterial level back to normal.
The demonstration of posthyperventilation apnea requires that the patient voluntarily take several deep breaths, so that it is useful in differential diagnosis of lethargic or confused patients, but not in cases of stupor or coma. One instructs the subject to take five deep breaths. No other instructions are given. It is useful for the examiner to place a hand on the patient’s chest, to make it easier later to detect when breathing has restarted, and to count the breaths. If the lungs function well, the maneuver usually lowers the arterial carbon dioxide by 8 to 14 torr. At the end of the deep breathing, wakeful patients without brain damage show little or no apnea (less than 10 seconds). However, in patients with forebrain impairment, the period of apnea may last from 12 to 30 seconds. The neural substrate that produces a continuous breathing pattern even in the absence of metabolic need is believed to include the same frontal pathways that regulate behavioral alterations of breathing patterns, as the continuous breathing pattern disappears with sleep, bilateral frontal lobe damage, or diffuse metabolic impairment of the hemispheres.
CHEYNE-STOKES RESPIRATION
Cheyne-Stokes respiration41 is a pattern of periodic breathing with phases of hyperpnea alternating regularly with apnea. The depth of respiration waxes from breath to breath in a smooth crescendo during onset of the hyperpneic phase and then, once a peak is reached, wanes in an equally smooth decrescendo until a period of apnea, usually from 10 to 20 seconds,

50 Plum and Posner’s Diagnosis of Stupor and Coma
A A

 B
B
C
D
E
B
C
D
1 min
E
Figure 2–5. Different abnormal respiratory patterns are associated with pathologic lesions (shaded areas) at various levels of the brain. Tracings by chest-abdomen pneumography, inspiration reads up. (A) Cheyne-Stokes respiration is seen with metabolic encephalopathies and with lesions that impair forebrain or diencephalic function. (B) Central neurogenic hyperventilation is most commonly seen in metabolic encephalopathies, but may rarely be seen in cases of high brainstem tumors. (C) Apneusis, consisting of inspiratory pauses, may be seen in patients with bilateral pontine lesions. (D) Cluster breathing and ataxic breathing are seen with lesions at the pontomedullary junction. (E) Apnea occurs when lesions encroach on the ventral respiratory group in the ventrolateral medulla bilaterally. (From Saper, C. Brain stem modulation of sensation, movement, and consciousness. Chapter 45 in: Kandel, ER, Schwartz, JH, Jessel, TM. Principles of Neural Science. 4th ed. McGraw-Hill, New York, 2000, pp. 871–909. By permission of McGraw-Hill.)
is reached. The hyperpneic phase usually lasts longer than the apneic phase (Figure 2–5).
This rhythmic alternation in Cheyne-Stokes respiration results from the interplay of normal brainstem respiratory reflexes.42–45 When the medullary chemosensory circuits sense adequate oxygen and carbon dioxide tension, they reduce the rate and depth of respiration, causing a gradual rise in arterial carbon dioxide tension. There is normally a short delay of a few seconds, representing the transit time for fresh blood from the lungs to reach the left heart and then the chemoreceptors in the carotid
artery and the brain. By the time the brain begins increasing the rate and depth of respiration, the alveolar carbon dioxide has reached even higher levels, and so there is a gradual ramping up of respiration as the brain sees a rising level of carbon dioxide, despite its additional efforts. By the time the brain begins to see a fall in carbon dioxide tension, the levels in the alveoli may be quite low. When blood containing this low level of carbon dioxide reaches the brain, respiration slows or may even cease, thus setting off another cycle. Hence, the periodic cycling is due to the delay (hys-
teresis) in the feedback loop between alveolar ventilation and brain chemoreceptor sensory responses.
The Cheyne-Stokes respiratory cycle is not usually seen in normal individuals because the circulatory delay between a change in alveolar blood gases and carbon dioxide tension in the brain is only a few seconds. Even as circulatory delay rises with cardiovascular or pulmonary disease, during waking the descending pathways that prevent posthyperventilation apnea also ensure the persistence of respiration even during periods of low metabolic need, thus damping the oscillations that produce CheyneStokes respiration. However, during sleep or with bilateral forebrain impairment, due either to a diffuse metabolic process such as uremia, hepatic failure, or bilateral damage such as cerebral infarcts or a forebrain mass lesion with diencephalic displacement, periodic breathing may emerge.43–45 In patients with heart failure, the transit time for blood from the lungs to reach the carotid and cerebral chemoreceptors can become so prolonged as to produce a Cheyne-Stokes pattern of respiration, even in the absence of forebrain impairment. Thus, Cheyne-Stokes respiration is mainly useful as a sign of intact brainstem respiratory reflexes in the patients with forebrain impairment, but cannot be interpreted in the presence of significant congestive heart failure.
HYPERVENTILATION IN
COMATOSE PATIENTS
Sustained hyperventilation is often seen in patients with impaired consciousness, but is usually a result of either hepatic coma or sepsis, conditions in which circulating chemical stimuli cause hyperpnea, or a metabolic acidosis, such as diabetic ketoacidosis (see Chapter 5). Other patients have meningitis caused either by infection or subarachnoid hemorrhage, which stimulates chemoreceptors in the brainstem,46 probably by altering CSF pH.
Some patients hyperventilate when intrinsic brainstem injury or subarachnoid hemorrhage or seizures cause neurogenic pulmonary edema.47 The ventilatory response is driven by pulmonary mechanosensory and chemosensory receptors. The pulmonary congestion lowers both the arterial carbon dioxide and the oxygen tension. Stimulation of pulmonary stretch re-
Examination of the Comatose Patient |
51 |
ceptors is apparently sufficient to cause reflex hyperpnea, as oxygen therapy sufficient to raise the arterial oxygen level does not always correct the overbreathing.
Another small group of patients has been identified who have hyperventilation associated with brainstem gliomas or lymphomas.48,49 These patients have spinal fluid that is acellular, but generally acidotic compared to arterial pH. In others, the lumbar CSF may have a normal pH, but it is believed that the tumor causes local lactic acidosis, which may trigger brain chemoreceptors to cause hyperventilation (Figure 2–5).
It is theoretically possible for an irritative lesion in the region of the parabrachial nucleus or other respiratory centers to produce hyperpnea.37 The diagnosis of such true ‘‘central neurogenic hyperventilation’’ requires that with the subject breathing room air, the blood gases show elevated arterial oxygen tension, decreased carbon dioxide tension, and an elevated pH. The cerebrospinal fluid likewise must show an elevated pH and be acellular. The respiratory changes must persist during sleep to eliminate psychogenic hyperventilation, and one must exclude the presence of stimulating drugs, such as salicylates, or disorders that stimulate respiration, such as hepatic failure or underlying systemic infection. Cases fulfilling all of these criteria have rarely been observed,50,51 and none that we are aware of has come to postmortem examination of the brain.
APNEUSTIC BREATHING
Apneusis is a respiratory pause at full inspiration. Fully developed apneustic breathing, with each cycle including an inspiratory pause, is rare in humans, but of considerable localizing value. Experiments in animals indicate that apneusis develops with injury to the pontine respiratory nuclei described above, and experi-
ence with rare human cases would support this view52,53 (see Figure 2–5).
Clinically, end-inspiratory pauses of 2 to 3 seconds usually alternate with end-expiratory pauses, and both are most frequently encountered in the setting of pontine infarction due to basilar artery occlusion. However, apneustic breathing may rarely be observed in metabolic encephalopathies, including hypoglycemia, anoxia, or meningitis. It is sometimes observed
52 Plum and Posner’s Diagnosis of Stupor and Coma
in cases of transtentorial herniation, as the brainstem dysfunction advances. At least one patient with apneusis due to a brainstem infarct responded to buspirone, a serotonin 1A receptor agonist.53
ATAXIC BREATHING
Irregular, gasping respiration implies damage to the respiratory rhythm generator at the preBo¨tzinger level of the upper medulla.30 This cell group can be specifically eliminated in experimental animals by the use of a toxin that binds to neurons that express NK-1 receptors. The resulting irregular, gasping breathing is eerily similar to humans with bilateral rostral medullary lesions, and it indicates that sufficient neurons survive in the medullary reticular formation to drive primitive ventilatory efforts, despite the loss of the neurons that cause smooth to-and-fro respiration.54 More complete bilateral lesions of the ventrolateral medullary reticular formation cause apnea, which is not compatible with life unless the patient is artificially ventilated (Figure 2–5).
A variety of intermediate types of breathing patterns are also seen with high medullary lesions. Some patients may breathe in irregular clusters or ratchet-like breaths separated by pauses. In other cases, particularly during intoxication with opiates or sedative drugs, the breathing may slow and decline in depth gradually until it fades into complete arrest.
There is a tendency in modern hospitals to intubate and ventilate patients with structural coma to protect the airway and permit treatment of respiratory failure. If the patient fights intubation or ventilation, paralytic drugs are often administered. This compromises the ability of the neurologist to assess brainstem reflexes, and in some cases may delay diagnosis and compromise care. Thus, it is important, whenever possible, to delay intubation until after the brief coma examination described here has been completed.
SLEEP APNEA AND ONDINE’S
CURSE
Obstructive sleep apnea is a common disorder in which the cross-section of the upper airway is anatomically narrow.55,56 During sleep, the muscles that keep the upper airway open, including the genioglossus muscle that pulls
the tongue forward, undergo a gradual loss of tone. This results in critical narrowing of the airway and the increased rate of movement of air tends to further reduce airway pressure, resulting in sudden closure. Liable to the disorder are obese patients, because deposition of fat in neck tissue reduces airway diameter; men, because the increased ratio of the length of the airway to its diameter predisposes to collapse; and middle aged or older patients, because muscle tone is more reduced during sleep with age. However, cases may occur in thin young adults, or even in children. Sleep apnea typically occurs in cycles lasting a few minutes each when the patient falls asleep, airway tone fails and an obstructive apnea occurs, blood oxygen levels fall, carbon dioxide rises, and the patient is aroused sufficiently to resume breathing. This cycle may be repeated many times over the course of a night. The fragmentation of sleep and intermittent hypoxia result in chronic daytime sleepiness and impairment of cognitive function, particularly vigilance.
Excessive drowsiness during the day and loud snoring at night may be the only clues. Lethargy or drowsiness due to neurologic injury may induce apneic cycles in a patient with obstructive sleep apnea. However, as the level of consciousness becomes more impaired, it may be difficult to achieve the periodic arousals necessary to resume breathing.
Other patients with pauses in ventilation have central sleep apnea. Most such patients have congestive heart failure, and the pauses are thought to be analogous to the periodic breathing that is seen in patients who develop Cheyne-Stokes respiration when they fall asleep.
Failure of automatic breathing is a rare condition, sometimes called Ondine’s curse, named after the mythologic wood nymph whose mortal lover lost autonomic functions whenever he went to sleep. In adults, Ondine’s curse is seen after lesions of the ventrolateral medullary chemosensory areas or bilateral damage to the descending pathways that control automatic respiration in the lateral columns of the
spinal cord (e.g., as a complication of cordotomy to relieve cancer pain).57–62 In children, it
is most frequently seen as a congenital condition in infants, sometimes in association with Hirschsprung’s disease, and either a neuroblastoma or pheochromocytoma, often associated
with a mutation in the PHOX2B gene.63 A variety of interventions have been successful, ranging from a rocking bed, which provides continuous somatic sensory and vestibular stimulation, to negative pressure ventilation, or even diaphragmatic pacing.64
YAWNING, HICCUPPING,
VOMITING
The neuronal pattern generators responsible for coordinating respiratory-related behaviors also are located in the ventrolateral medulla, in close proximity to the nucleus ambiguus.65 Yawning is a motor pattern that involves deep inspiration associated with wide opening of the jaw and generalized muscle stretching.66,67 It is seen even in patients who are locked in, and hence is apparently organized at a medullary level. Yawning may improve the compliance of the lungs and chest wall, but its function is not understood. It may be seen in lethargic patients, but yawning is also seen in complex partial seizures emanating from the medial temporal lobe, and is not of great localizing value.
Hiccups occur in patients with abdominal
or subphrenic pathology (e.g., pancreatic cancer) that impinges upon the vagus nerve.68,69
Dexamethasone may induce hiccups; the mechanism is unknown.70 Hiccups occasionally occur with lesions in the medullary tegmentum, including neoplasms, infarction, hematomas, infections, or syringobulbia. Because stuporous patients with intracranial mass lesions are often treated with corticosteroids to reduce brain edema, it may be difficult to determine whether pressure on the floor of the fourth ventricle from the mass lesion or the treatment with corticosteroids is causing the hiccups.71 Pathologic hiccupping is peculiarly more common in men; in a study of 220 patients at the
Mayo Clinic with pathologic hiccupping, all but 39 were men.72
The hiccup reflex consists of a spasmodic burst of inspiratory activity, followed 35 milliseconds later by abrupt glottic closure, so that the ventilatory effect is negligible. On the other hand, if the airway is kept open artificially (e.g., by tracheostomy), the inrush of air can be sufficient to hyperventilate the patient. As an example, one patient in New York Hospital with a low brainstem infarct and tracheostomy maintained his total ventilation for several days by hiccup alone.
Examination of the Comatose Patient |
53 |
Pathologic hiccups are difficult to treat.73 A number of drugs and physical approaches have been tried, most of which do not work well. Agents used to treat hiccups include phenothiazines, calcium channel blockers, baclofen, and anticonvulsants, gabapentin being the most recent.74 In steroid-induced hiccups, decreasing the dose usually reduces the hiccups.73
Vomiting is a reflex response involving coordinated somatomotor (posture, abdominal muscle contraction), gastrointestinal (reversal of peristalsis), and respiratory (retching, breath holding) components that are coordinated by neurons in the ventrolateral medullary tegmentum near the compact portion of the nucleus ambiguus. The vomiting reflex may be triggered by vagal afferents75,76 or by chemosensory neurons in the area postrema, a small group of nerve cells that sits atop the nucleus of the solitary tract in the floor of the fourth ventricle, just at the level of the obex.77
In patients with impaired consciousness, vomiting is frequently due to lesions involving the lateral pons or medulla, causing vestibular imbalance. It occasionally occurs in patients with irritative lesions limited to the region of the nucleus of the solitary tract.77 Such vomiting is typically preceded by intense nausea. More commonly, however, vomiting is due to a sudden increase in intracranial pressure, such as occurs in subarachnoid hemorrhage. The pressure wave may stimulate the emetic response directly by pressure on the floor of the fourth ventricle, resulting in sudden, ‘‘projectile’’ vomiting, without warning. This type of vomiting is particularly common in children with posterior fossa tumors. It is also seen in adults with brain tumor, who hypoventilate during sleep, resulting in cerebral vasodilation. The small increase in intravascular blood volume, in a patient whose intracranial pressure is already elevated, may cause a sharp increase in intracranial pressure (see Chapter 3), resulting in onset of an intense headache that may waken the patient, followed shortly thereafter by sudden projectile vomiting. Children with posterior fossa tumors may simply vomit without headache.
Vomiting is also commonly seen in patients with brain tumors during chemotherapy or even radiation therapy. Tissue injury, particularly in the gut, may release emetic hormones, such as glucagon-like peptide-1 (GLP-1). GLP- 1 is detected by neurons in the area postrema,
54 Plum and Posner’s Diagnosis of Stupor and Coma
and it can induce a vomiting reflex.78 The area postrema contains both dopaminergic and serotoninergic neurons, and the latter produce emesis primarily by means of contacting 5HT3 receptors.77 Hence, drugs that block dopamine D2 receptors (e.g., chlorpromazine, metoclopramide) or serotonin 5HT3 receptors (ondansetron) are effective antiemetics.
PUPILLARY RESPONSES
The pupillary light reflex is one of the most basic and easily tested nervous system responses. It is controlled by a complex balance of sympathetic (pupillodilator) and parasympathetic (pupilloconstrictor) pathways (see Figure 2– 6). The anatomy of these pathways is closely intertwined with the components of the ascending arousal system. In addition, the pupillary pathways are among the most resistant to metabolic insult. Hence, abnormalities of pupillary responses are of great localizing value in diagnosing the cause of stupor and coma, and the pupillary light reflex is the single most important physical sign in differentiating metabolic from structural coma.
Examine the Pupils and
Their Responses
If possible, inquire if the patient has suffered eye disease or uses eyedrops. Observe the pupils in ambient light; if room lights are bright and pupils are small, dimming the light may make it easier to see the pupillary responses. They should be equal in size and about the same size as those of normal individuals in the same light (8% to 18% of normal individuals have anisocoria greater than 0.4 mm). Unequal pupils can result from sympathetic paralysis making the pupil smaller or parasympathetic paralysis making the pupil larger. If one suspects sympathetic paralysis (see Horner’s syndrome, page 58), dim the lights in the room, allowing the normal pupil to dilate and thus bringing out the pupillary inequality. Unless there is specific damage to the pupillary system, pupils of stuporous or comatose patients are usually smaller than normal pupils in awake subjects. Pupillary responses must be examined with a bright light. The eyelids can be held
open while the light from a bright flashlight illuminates each pupil. Shining the light into one pupil should cause both pupils to react briskly and equally. Because the pupils are often small in stuporous or comatose patients and the light reflex may be through a small range, one may want to view the pupil through the bright light of an ophthalmoscope using a plus 20 lens or through the lens of an otoscope. Most pupillary responses are brisk, but a tonic pupil may react slowly, so the light should illuminate the eye for at least 10 seconds. Moving the light from one eye to the other may result in constriction of both pupils when the light is shined into the first eye, but paradoxically pupillary dilation when the light is shined in the other eye. This aberrant pupillary response results from damage to the retina or optic nerve on the side on which the pupil dilates (relative afferent pupillary defect [RAPD]).79
One of the most ominous signs in neurology is a unilateral dilated and unreactive pupil. In a comatose patient, this usually indicates oculomotor nerve compromise either by a posterior communicating artery aneurysm or by temporal lobe herniation (see oculomotor responses, page 60). However, the same finding can be mimicked by unilateral instillation of atropinelike eye drops. Occasionally this happens by accident, as when a patient who is using a scopolamine patch to avert motion sickness inadvertently gets some scopolamine onto a finger when handling the patch, and then rubs the eye; however, it is also seen in cases of factitious presentation. Still other times, unilateral pupillary dilation may occur in the setting of ciliary ganglion dysfunction from head or facial trauma. In most of these cases there is a fracture in the posterior floor of the orbit that interrupts the fibers of the inferior division of the oculomotor nerve.80 Injury to the third nerve can be distinguished from atropinic blockade at the bedside by instilling a dilute solution of pilocarpine into the eye (see pharmacology, page 56). The denervated pupil will respond briskly, whereas the one that is blocked by atropine will not.81
Once both the ipsilateral and consensual pupillary light reflexes have been noted, the next step is to induce a ciliospinal reflex.10 This can be done by pinching the skin of the neck or the face. The pupils should dilate 1 to 2 mm bilaterally. This reflex is an example of a spinobulbospinal response (i.e., the pain stimulus

arises from the trigeminal or spinal dorsal horn, must ascend to brainstem autonomic control areas, and then descend again to the C8-T2 sympathetic preganglionic neurons). A normal ciliospinal response ensures integrity of these circuits from the lower brainstem to the spinal cord, thus usually placing the lesion in the rostral pons or higher.
Pathophysiology of Pupillary
Responses: Peripheral Anatomy
of the Pupillomotor System
The pupil is a hole in the iris; thus, change in pupillary diameter occurs when the iris contracts or expands. The pupillodilator muscle is a set of radially oriented muscle fibers, running from the edge of the pupil to the limbus (outer edge) of the iris. When these muscles contract, they open the pupil in much the way a drawstring pulls up a curtain. The pupillodilator muscles are innervated by sympathetic ganglion cells in the superior cervical ganglion. These
Examination of the Comatose Patient |
55 |
axons pass along the internal carotid artery, joining the ophthalmic division of the trigeminal nerve in the cavernous sinus and accompanying it through the superior orbital fissure, into the orbit. Sympathetic input to the lid retractor muscle takes a similar course, but sympathetic fibers from the superior cervical ganglion that control facial sweating travel along the external carotid artery. Hence, lesions of the ascending cervical sympathetic chain up to the superior cervical ganglion typically give rise to Horner’s syndrome (ptosis, miosis, and facial anhydrosis). However, lesions along the course of the internal carotid artery may give only the first two components of this syndrome (Raeder’s paratrigeminal syndrome). The sympathetic preganglionic neurons for pupillary control are found in the intermediolateral column of the first three thoracic segments. Hence, lesions of those roots, or of the ascending sympathetic trunk between T1 and the superior cervical ganglion, may also cause a Horner’s syndrome with, depending on the exact site of the lesion, anhydrosis of the ipsilateral face or the face and arm.
A |
|
B |
|
|
|
|
|
Pupilloconstrictor |
|
|
|
|
|
|
muscle in the iris |
|
|
|
|
|
Retinal |
|
|
Short |
|
|
|
ganglion cell |
|
|
|
|
|
|
|
Pupillodilator |
ciliary |
Ciliary |
|
|
|
|
Ciliary |
|
|
|||
|
muscle in the iris |
nerve |
ganglion |
|
|
|
|
ganglia |
|
|
|||
|
|
|
|
|
|
|
|
|
|
|
|
|
Hypothalamus |
|
|
|
Long |
|
Ophthalmic |
Internal |
|
|
|
ciliary nerve |
division |
carotid |
|
|
|
|
|
|
trigeminal |
artery |
|
|
|
|
|
nerve |
|
|
|
|
|
|
|
3rd |
|
III |
|
|
|
Superior |
neuron |
|
|
|
|
cervical |
|
|
|
nerve |
|
|
|
|
|
|
|
|
|
sympathetic |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
ganglion |
|
|
|
|
|
|
|
T1 |
LGN |
|
|
|
|
|
T2 |
|
|
|
|
|
|
|
MLF |
|
Edinger-Westphal |
|
|
|
T3 |
|
|
|
|
|
||
Olivary pretectal |
|
nucleus |
|
|
|
|
nucleus
Figure 2–6. Two summary drawings indicating the (A) parasympathetic pupilloconstrictor pathways and (B) sympathetic pupillodilator pathways. LGN, lateral geniculate nucleus; MLF, medial longitudinal fasciculus. (From Saper, C. Brain stem modulation of sensation, movement, and consciousness. Chapter 45 in: Kandel, ER, Schwartz, JH, Jessel, TM. Principles of Neural Science. 4th ed. McGraw-Hill, New York, 2000, pp. 871–909. By permission of McGraw-Hill.)