

Книги по МРТ КТ на английском языке / PLUM AND POSNER S DIAGNOSIS OF STUPOR AND COM-1
.pdf86 Plum and Posner’s Diagnosis of Stupor and Coma
115.Fisher CM. Some neuro-ophthalmological observations. J Neurol Neurosurg Psychiatry 30 (5), 383– 392, 1967.
116.Chung CS, Caplan LR, Yamamoto Y, et al. Striatocapsular haemorrhage. Brain 123 (Pt 9), 1850–1862, 2000.
117.Baloh RW, Furman JM, Yee RD. Dorsal midbrain syndrome: clinical and oculographic findings. Neurology 35 (1), 54–60, 1985.
118.Choi KD, Jung DS, Kim JS. Specificity of ‘‘peering at the tip of the nose’’ for a diagnosis of thalamic hemorrhage. Arch Neurol 61, 417–422, 2004.
119.Litvan I, Jankovic J, Goetz CG, et al. Accuracy of the clinical diagnosis of postencephalitic parkinsonism: a clinicopathologic study. Eur J Neurol 5 (5), 451–457, 1998.
120.Jhee SS, Zarotsky V, Mohaupt SM, et al. Delayed onset of oculogyric crisis and torticollis with intramuscular haloperidol. Ann Pharmacother 37 (10), 1434–1437, 2003.
121.Pannullo SC, Reich JB, Krol G, et al. MRI changes in intracranial hypotension. Neurology 43, 919–926, 1993.
122.Keane JR. Alternating skew deviation: 47 patients. Neurology 35 (5), 725–728, 1985.
123.Brandt TH, Dieterich M. Different types of skew deviation. J Neurol Neurosurg Psychiatry 54 (6), 549– 550, 1991.
124.Keane JR. Ocular skew deviation. Analysis of 100 cases. Arch Neurol 32 (3), 185–190, 1975.
125.Smith JL, David NJ, Klintworth G. Skew deviation. Neurology 14, 96–105, 1964.
126.Johkura K, Komiyama A, Tobita M, et al. Saccadic ping-pong gaze. J Neuroophthalmol 18, 43–46, 1998.
127.Daroff RB, Hoyt WF. Supranuclear disorders of ocular control systems in man: clinical, anatomical and physiological correlations. In: Bach-y-Rita P, Collins CC, Hyde JE, eds. The Control of Eye Movements. New York: Academic Press, pp 175–235, 1971.
128.Ochs AL, Stark L, Hoyt WF, et al. Opposed adducting saccades in convergence-retraction nystagmus: a patient with sylvian aqueduct syndrome. Brain 102 (3), 497–508, 1979.
129.Fischer CM. Ocular bobbing. Arch Neurol 11, 543– 546, 1964.
130.Rosenberg ML. Spontaneous vertical eye movements in coma. Ann Neurol 20 (5), 635–637, 1986.
131.Herishanu YO, Abarbanel JM, Frisher S, et al. Spontaneous vertical eye movements associated with pontine lesions. Isr J Med Sci 27 (6), 320–324, 1991.
132.Lourie H. Seesaw nystagmus. Case report elucidating the mechanism. Arch Neurol 9, 531–533, 1963.
133.Sano K, Sekino H, Tsukamoto Y, et al. Stimulation and destruction of the region of the interstitial nucleus in cases of torticollis and see-saw nystagmus. Confin Neurol 34 (5), 331–338, 1972.
134.Keane JR. Intermittent see-saw eye movements. Report of a patient in coma after hyperextension head injury. Arch Neurol 35 (3), 173–174, 1978.
135.Schott JM, Rossor MN. The grasp and other primitive reflexes. J Neurol Neurosurg Psychiatry 74 (5), 558–560, 2003.
136.Jacobs L, Gossman MD. Three primitive reflexes in normal adults. Neurology 30 (2), 184–188, 1980.
137.De RE, Barbieri C. The incidence of the grasp reflex following hemispheric lesion and its relation to frontal damage. Brain 115 (Pt 1), 293–313, 1992.
138.Greenberg DA, Simon RP. Flexor and extensor postures in sedative drug-induced coma. Neurology 32 (4), 448–451, 1982.
139.Jennett B, Teasdale G. Aspects of coma after severe head injury. Lancet 1(8017), 878–881, 1977.
140.Sherrington CS. Cataleptoid reflexes in the monkey. Proc Royal Soc Lond 60, 411–414, 1897.
141.Kirk MM, Hoogwerf BJ, Stoller JK. Reversible decerebrate posturing after profound and prolonged hypoglycemia. Cleve Clin J Med 58 (4), 361–363, 1991.
142.Conomy JP, Swash M. Reversible decerebrate and decorticate postures in hepatic coma. N Engl J Med 278 (16), 876–879, 1968.
143.Strauss GI, Moller K, Larsen FS, et al. Cerebral glucose and oxygen metabolism in patients with fulminant hepatic failure. Liver Transpl 9 (12), 1244– 1252, 2003.
144.Kosaka Y, Tanaka K, Sawa H, et al. Acid-base disturbance in patients with fulminant hepatic failure. Gastroenterol Jpn 14(1), 24–30, 1979.
145.Krapf R, Caduff P, Wagdi P, et al. Plasma potassium response to acute respiratory alkalosis. Kidney Int 47 (1), 217–224, 1995.
146.Spector RH, Davidoff RA, Schwartzman RJ. Phenytoin-induced ophthalmoplegia. Neurology 26 (11), 1031–1034, 1976.
147.Pulst SM, Lombroso CT. External ophthalmoplegia, alpha and spindle coma in imipramine overdose: case report and review of the literature. Ann Neurol 14(5), 587–590, 1983.
148.Odaka M, Yuki N, Yamada M, et al. Bickerstaff’s brainstem encephalitis: clinical features of 62 cases and a subgroup associated with Guillain-Barre syndrome. Brain 126 (Pt 10), 2279–2290, 2003.
149.Ragosta K. Miller Fisher syndrome, a brainstem encephalitis, mimics brain death. Clin Pediatr (Phila) 32 (11), 685–687, 1993.
150.Dhiravibulya K, Ouvrier R, Johnston I, et al. Benign intracranial hypertension in childhood: a review of 23 patients. J Paediatr Child Health 27 (5), 304–307, 1991.
151.Thomke F, Mika-Gruttner A, Visbeck A, et al. The risk of abducens palsy after diagnostic lumbar puncture. Neurology 54 (3), 768–769, 2000.
152.Speer C, Pearlman J, Phillips PH, et al. Fourth cranial nerve palsy in pediatric patients with pseudotumor cerebri. Am J Ophthalmol 127 (2), 236–237, 1999.
153.Malouf R, Brust JC. Hypoglycemia: causes, neurological manifestations, and outcome. Ann Neurol 17(5), 421–430, 1985.
154.Vaughan CJ, Delanty N. Hypertensive emergencies. Lancet 356 (9227), 411–417, 2000.
155.Mitchell P, Wilkinson ID, Hoggard N, et al. Detection of subarachnoid haemorrhage with magnetic resonance imaging. J Neurol Neurosurg Psychiatry 70, 205–211, 2001.
156.Lin A, Ross BD, Harris K, et al. Efficacy of proton magnetic resonance spectroscopy in neurological diagnosis and neurotherapeutic decision making. NeuroRx 2(2), 197–214, 2005.
157.Guillevin R, Vallee JN, Demeret S, et al. Cerebral fat embolism: Usefulness of magnetic resonance spectroscopy. Ann Neurol 57, 434–439, 2005.
158.Schoning M, Scheel P, Holzer M, et al. Volume measurement of cerebral blood flow: assessment of cerebral circulatory arrest. Transplantation 80 (3), 326–331, 2005.
159.Dominguez-Roldan JM, Garcia-Alfaro C, JimenezGonzalez PI, et al. Brain death due to supratentorial masses: diagnosis using transcranial Doppler sonography. Transplant Proc 36 (10), 2898–2900, 2004.
160.Wojner-Alexandrov AW, Alexandrov AV, Rodriguez D, et al. Houston paramedic and emergency stroke treatment and outcomes study (HoPSTO). Stroke 36 (7), 1512–1518, 2005.
161.Panerai RB, Kerins V, Fan L, et al. Association between dynamic cerebral autoregulation and mortality in severe head injury. Br J Neurosurg 18 (5), 471–479, 2004.
162.Droste DW, Metz RJ. Clinical utility of echocontrast agents in neurosonology. Neurol Res 26 (7), 754– 759, 2004.
Examination of the Comatose Patient |
87 |
163.Brenner RP. The interpretation of the EEG in stupor and coma. Neurologist 11(5), 271–284, 2005.
164.Kaplan PW. Assessing the outcomes in patients with nonconvulsive status epilepticus: nonconvulsive status epilepticus is underdiagnosed, potentially overtreated, and confounded by comorbidity. J Clin Neurophysiol 16 (4), 341–352, discussion 353, 1999.
165.Brenner RP. Is it status? Epilepsia 43 (Suppl 3), 103–113, 2002.
166.Towne AR, Waterhouse EJ, Boggs JG, et al. Prevalence of nonconvulsive status epilepticus in comatose patients. Neurology 54, 340–345, 2000.
167.Burneo JG, Knowlton RC, Gomez C, et al. Confirmation of nonconvulsive limbic status epilepticus with the sodium amytal test. Epilepsia 44, 1122– 1126, 2003.
168.Kaplan PW. The clinical features, diagnosis, and prognosis of nonconvulsive status epilepticus. Neurologist 11(6), 348–361, 2005.
169.Fischer C, Luaute´ J, Adeleine P, et al. Predictive value of sensory and cognitive evoked potentials for awakening from coma. Neurology 63, 669–673, 2004.

Chapter 3
Structural Causes of
Stupor and Coma
COMPRESSIVE LESIONS AS A CAUSE OF COMA
COMPRESSIVE LESIONS MAY DIRECTLY
DISTORT THE AROUSAL SYSTEM
Compression at Different Levels
of the Central Nervous System Presents
in Distinct Ways
The Role of Increased Intracranial Pressure
in Coma
The Role of Vascular Factors and Cerebral
Edema in Mass Lesions
HERNIATION SYNDROMES:
INTRACRANIAL SHIFTS IN
THE PATHOGENESIS OF COMA
Anatomy of the Intracranial Compartments
Patterns of Brain Shifts That Contribute
to Coma
Clinical Findings in Uncal Herniation
Syndrome
Two major classes of structural brain injuries cause coma (Table 3–1): (1) Compressive lesions may impair consciousness either by directly compressing the ascending arousal system or by distorting brain tissue so that it moves out of position and secondarily compresses components of the ascending arousal system or its forebrain targets (see herniation syndromes, page 95). These processes include a wide range of space-occupying lesions such as tumor, hematoma, and abscess. (2) Destructive lesions
Clinical Findings in Central Herniation
Syndrome
Clinical Findings in Dorsal Midbrain
Syndrome
Safety of Lumbar Puncture in Comatose
Patients
False Localizing Signs in the Diagnosis
of Structural Coma
DESTRUCTIVE LESIONS AS A CAUSE OF COMA
DIFFUSE, BILATERAL CORTICAL DESTRUCTION
DESTRUCTIVE DISEASE OF
THE DIENCEPHALON
DESTRUCTIVE LESIONS OF
THE BRAINSTEM
cause coma by direct damage to the ascending arousal system or its forebrain targets. To cause coma, lesions of the diencephalon or brainstem must be bilateral, but can be quite focal if they damage the ascending activating system near the midline in the midbrain or caudal diencephalon; cortical or subcortical damage must be both bilateral and diffuse. Processes that may cause these changes include tumor, hemorrhage, infarct, trauma, or infection. Both destructive and compressive lesions may cause
88

Structural Causes of Stupor and Coma |
89 |
Table 3–1 Sites and Representative Causes of Structural Lesions That Can Cause Coma
Compressive |
Destructive |
|
|
Cerebral |
Cerebral hemisphere |
Bilateral subdural hematomas |
Cortex (e.g., acute anoxic injury) |
Diencephalon |
Subcortical white matter |
(e.g., delayed anoxic injury) |
|
Thalamus (e.g., hemorrhage) |
Diencephalon |
Hypothalamus (e.g., pituitary tumor) |
|
Brainstem |
Thalamus (e.g., infarct) |
|
|
Midbrain (e.g., uncal herniation) |
Brainstem |
Cerebellum (e.g., tumor, |
Midbrain, pons (e.g., infarct) |
hemorrhage, abscess) |
|
|
|
additional compression by producing brain edema.
Most compressive lesions are treated surgically, whereas destructive lesions are generally treated medically. This chapter describes the pathophysiology and general approach to patients with structural lesions of the brain, first considering compressive and then destructive lesions. Chapter 4 deals with some of the specific causes of coma outlined in Table 3–1.
Chapter 2 has described some of the physical findings that distinguish structural from nonstructural causes of stupor and coma. The physician must first decide whether the patient is indeed stuporous or comatose, distinguishing those patients who are not in coma but suffer from abulia, akinetic mutism, psychologic unresponsiveness, or the locked-in state from those truly stuporous or comatose (see Chapter 1). This is usually relatively easily done during the course of the initial examination. More difficult is distinguishing structural from metabolic causes of stupor or coma. As indicated in Chapter 2, if the structural cause of coma involves the ascending arousal system in the brainstem, the presence of focal findings usually makes the distinction between metabolic and structural coma easy. However, when the structural disease involves the cerebral cortex diffusely or the diencephalon bilaterally, focal signs are often absent and it may be difficult to distinguish structural from metabolic coma. Compressive lesions that initially do not cause focal signs eventually do so, but by then coma
may be irreversible. Thus, if there is any question about the distinction between structural and metabolic coma, immediately after stabilizing the patient, an imaging study (usually a computed tomography [CT] scan but, if available, a magnetic resonance imaging [MRI] scan) must be obtained to rule out a mass lesion that may be surgically remediable. Identifying surgically remediable lesions that have not yet caused focal findings gives the physician time to stabilize the patient and investigate other additional nonstructural causes of coma. The time, however, is short and should be counted in minutes rather than hours or days. If focal findings are already present, efforts to decrease intracranial pressure (ICP), including hyperventilation and hyperosmolar agents and often administration of corticosteroids (Chapter 7), should be instituted before sending the patient for imaging.
COMPRESSIVE LESIONS AS A CAUSE OF COMA
Compressive lesions may impair consciousness in a number of critical ways: (1) by directly distorting the arousal system or its forebrain targets; (2) by increasing ICP diffusely to the point of impairing global cerebral blood flow; (3) by distorting tissue to the point of causing local ischemia; (4) by causing edema, thus further distorting neural tissue; or (5) by causing tissue shifts (herniations). Understanding the
90 Plum and Posner’s Diagnosis of Stupor and Coma
anatomy and pathophysiology of each of these processes is critical in evaluating patients in coma.
COMPRESSIVE LESIONS
MAY DIRECTLY DISTORT
THE AROUSAL SYSTEM
Compression at key levels of the brain may cause coma by exerting pressure upon the structures of the arousal system. The mechanism by which local pressure may impair neuronal function is not entirely understood. However, neurons are dependent upon axonal transport to supply critical proteins and mitochondria to their terminals, and to transport used or damaged cellular components back to the cell body for destruction and disposal. Even a loose ligature around an axon causes damming of axon contents on both sides of the stricture, due to impairment of both anterograde and retrograde axonal flow, and results in impairment of axonal function. Perhaps the clearest example of this relationship is provided by the optic nerve in patients with papilledema (see section on increased ICP, page 91). When a compressive lesion results in displacement of the structures of the arousal system, consciousness may become impaired, as described in the sections below.
Compression at Different Levels
of the Central Nervous System
Presents in Distinct Ways
When a cerebral hemisphere is compressed by a lesion such as a subdural hematoma, tumor, or abscess that grows slowly over a long period of time, it may reach a relatively large size with little in the way of local signs that can help identify the diagnosis. The tissue in the cerebral hemispheres can absorb a surprising amount of distortion and stretching, as long as the growth of the mass can be compensated for by displacing cerebrospinal fluid (CSF) from the ventricles in that hemisphere. However, when there is no further room in the hemisphere to expand, even a small amount of growth can only be accommodated by compressing the diencephalon and midbrain either laterally across the midline or downward. In such patients, the
impairment of consciousness correlates with the displacement of the diencephalon and upper brainstem in a lateral or caudal direction.1 Hence, when a patient with a hemispheric lesion reaches the point of impairment of consciousness, there is very little time left to intervene before the brain is irreparably injured.
The diencephalon may also be compressed by a mass lesion in the thalamus itself (generally a tumor or a hemorrhage) or a mass in the suprasellar cistern (typically a craniopharyngioma, a germ cell tumor, or suprasellar extension of a pituitary adenoma; see Chapter 4). In addition to causing impairment of consciousness, suprasellar tumors typically cause visual field deficits, classically a bitemporal hemianopsia, although a wide range of optic nerve or tract injuries may also occur. If a suprasellar tumor extends into the cavernous sinus, there may be injury to the cranial nerves that supply the ocular muscles (III, IV, VI) and the ophthalmic division of the trigeminal nerve (V1). On occasion, these tumors may also cause endocrine dysfunction. If they damage the pituitary stalk, they may cause diabetes insipidus or panhypopituitarism. In women, the presence of a pituitary tumor is often heralded by galactorrhea and amenorrhea, as prolactin is the sole anterior pituitary hormone under negative regulation, and it is typically elevated when the pituitary stalk is damaged.
The dorsal midbrain may be compressed by a tumor in the pineal region. Pineal mass lesions may be suprasellar germinomas or other germ cell tumors (embryonal cell carcinoma, teratocarcinoma) that occur along the midline, or pineal masses including pinealcytoma or pineal astrocytoma. Pineal masses compress the pretectal area as well. Thus, in addition to causing impairment of consciousness, they produce diagnostic neuro-ophthalmologic signs including fixed, slightly enlarged pupils; impairment of voluntary vertical eye movements (typically elevation is impaired earlier and more severely than depression) and convergence; and convergence nystagmus and sometimes retractory nystagmus (Parinaud’s syndrome; see page 110).2 Hemorrhage into the pulvinar of the thalamus, which overlies the pretectal area and dorsal midbrain, may sometimes produce a similar constellation of signs.
Posterior fossa compressive lesions most often originate in the cerebellum, including tumors, hemorrhages, infarctions, or abscesses, although
occasionally extra-axial lesions, such as a subdural or epidural hematoma, may have a similar effect. Tumors of the cerebellum include the full range of primary and metastatic brain tumors (Chapter 4), as well as juvenile pilocytic astrocytomas and medulloblastomas in children and hemangioblastoma in patients with von Hippel-Lindau syndrome.
A cerebellar mass causes coma by direct compression of the brainstem, which may also cause the brainstem to herniate upward through the tentorial notch. As the patient loses consciousness, there is a pattern of pontine level dysfunction, with small reactive pupils, impairment of vestibulo-ocular responses (which may be asymmetric), and decerebrate motor responses.3,4 Because the base of the pons is farthest from the cerebellum, motor signs (e.g., upgoing toes) are usually a relatively late finding, and suggest instead an intrinsic brainstem mass. With upward pressure on the midbrain, the pupils become asymmetric or unreactive. If vestibuloocular responses were not previously impaired by pontine compression, vertical eye movements may be lost.
Cerebellar mass lesions may also cause coma by compressing the fourth ventricle to the point where it impairs flow of CSF. This causes acute hydrocephalus and rapidly increasing ICP (see page 147). The onset of obstruction of the fourth ventricle is typically heralded by nausea and sometimes sudden, projectile vomiting. There may also be a history of ataxia, vertigo, neck stiffness, and eventually respiratory arrest as the cerebellar tonsils are impacted upon the lip of the foramen magnum. If the compression develops slowly (i.e., over more than 12 hours), there may also be papilledema. Because cerebellar masses may cause acute obstruction of the fourth ventricle by expanding by only a few millimeters in diameter, they are potentially very dangerous.
On occasion, impairment of consciousness may occur as a result of a mass lesion directly compressing the brainstem. These are more commonly intrinsic masses, such as an abscess or a hemorrhage, in which case it is difficult to determine how much of the impairment is due to compression as opposed to destruction. Occasionally, a mass lesion of the cerebellopontine angle, such as a vestibular schwannoma, meningioma, or cholesteatoma, may compress the brainstem. However, these are usually slow processes and the mass may reach a very large
Structural Causes of Stupor and Coma |
91 |
size and often causes signs of local injury before consciousness is impaired.
The Role of Increased Intracranial
Pressure in Coma
A key and often misunderstood point is that increases in ICP are withstood remarkably well by the brain, as long as they progress relatively slowly. In patients with chronic elevation of CSF pressure, such as those with pseudotumor cerebri, there is little evidence of brain dysfunction, even when CSF pressures reach 600 mm of water or greater. The chief problems induced by increased ICP are papilledema and headache, until the pressure gets high enough to impair cerebral blood flow.
Papilledema is due to the pressure differential applied to the optic nerve by the increase in ICP. Retinal ganglion cells within the eye are subject to intraocular pressure, typically in the same range as normal CSF pressure. Their axons leave the eye through the optic disk and travel to the brain via the optic nerve. Axoplasm flows from the retinal ganglion cell bodies in the eye, down the axon and through the optic disc. Similarly, the retinal veins within the eye are subject to intraocular pressure. They also leave through the optic disc and run along the optic nerve. The optic nerve in turn is surrounded by a dural and arachnoid sleeve, which contains CSF that communicates with the CSF in the subarachnoid space around the brain.5 The optic disk itself is composed of a dense fibrous network forming a cribriform (from the Latin for sieve) plate that acts as a pressure fitting, so that the optic nerve and retinal vein are exposed to intraocular pressure on one side of the disk and to ICP on the other side.
Normally, axonal transport proceeds unimpeded and the retinal veins show normal venous pulsations, as there is little, if any, pressure differential between the two compartments. As ICP rises above systemic venous pressure, retinal venous pulsations are damped or eliminated as an early feature of papilledema. The retinal veins become larger and more numerous appearing, because increased venous pressure causes smaller veins to become more noticeable on funduscopy. Thus, the presence of retinal venous pulsations is a good but not invariable sign of normal ICP, and engorgement of retinal veins is a reliable early sign of
92 Plum and Posner’s Diagnosis of Stupor and Coma
increased ICP.6,7 A second consequence of increased ICP is that axoplasmic flow is impaired (as if a loose ligature had been tied around the nerve), and there is buildup of axoplasm on the retinal side of the disk. The swollen optic axons obscure the disk margins, beginning at the superior and inferior poles, then extending laterally and finally medially.8 The size of the optic disk increases, and this can be mapped as a larger ‘‘blind spot’’ in the visual field. Some patients even complain of a visual scotoma in this area. If ICP is increased sufficiently, the ganglion cells begin to fail from the periphery of the retina in toward the macula. This results in a concentric loss of vision.
Because papilledema reflects the backpressure on the optic nerves from increased ICP, it is virtually always bilateral. A rare exception occurs when the optic nerve on one side is itself compressed by a mass lesion (such as an olfactory groove meningioma), thus resulting in optic atrophy in one eye and papilledema in the other eye (the Foster Kennedy syndrome). On the other hand, optic nerve injury at the level of the optic disk, either due to demyelinating disease or vascular infarct of the vasa nervorum (anterior ischemic optic neuropathy), can also block axonal transport and venous return, due to retrobulbar swelling of the optic nerve.9 The resulting papillitis can look identical to papilledema but is typically unilateral, or at least does not involve the optic nerves simultaneously. In addition, papillitis is usually accompanied by the relatively rapid onset of visual loss, particularly focal loss called a scotoma, so the clinical distinction is usually clear.
The origin of headache in patients with increased ICP is not understood. CSF normally leaves the subarachnoid compartment mainly by resorption at the arachnoid villi.10 These structures are located along the surface of the superior sagittal sinus, and they consist of invaginations of the arachnoid membrane into the wall of the sinus. CSF is taken up from the subarachnoid space by endocytosis into vesicles, the vesicles are transported across the arachnoid epithelial cells, and then their contents are released by exocytosis into the venous sinus. Imbalance in the process of secretion and resorption of CSF occurs in cases of CSFsecreting tumors as well as in pseudotumor cerebri. In both conditions, very high levels of CSF pressure, in excess of 600 mm of water, may be achieved, but rather little in the way of
brain dysfunction occurs, other than headache. Experimental infusion of artificial CSF into the subarachnoid space, to pressures as high as 800 or even 1,000 mm of water, also does
not cause cerebral dysfunction and, curiously, often does not cause headache.11,12 However,
conditions that cause diffusely increased ICP such as pseudotumor cerebri usually do cause headache,13 suggesting that they must cause some subtle distortion of pain receptors in the cerebral blood vessels or the meninges.14
On the other hand, when there is obstruction of the cerebral venous system, increased ICP is often associated with signs of brain dysfunction as well as severe headache. The headache is localized to the venous sinus that is obstructed (superior sagittal sinus headache is typically at the vertex of the skull, whereas lateral sinus headache is usually behind the ear on the affected side). The headache in these conditions is thought to be due to irritation and local distortion of the sinus itself. Brain dysfunction is produced by back-pressure on the draining veins that feed into the sinus, thus reducing the perfusion pressure of the adjacent areas of the brain, to the point of precipitating venous infarction (see page 154). Small capillaries may be damaged, producing local hemorrhage and focal or generalized seizures. Superior sagittal sinus thrombosis produces parasagittal ischemia in the hemispheres, causing lower extremity paresis. Lateral sinus thrombosis typically causes infarction in the inferior lateral temporal lobe, which may produce little in the way of signs, other than seizures.
The most important mechanism by which diffusely raised ICP can cause symptoms is by impairment of the cerebral arterial supply. The brain usually compensates for the increased ICP by regulating its blood supply as described in Chapter 2. However, as ICP reaches and exceeds 600 mm of water, the back-pressure on cerebral perfusion reaches 45 to 50 mm Hg, which becomes a major hemodynamic challenge. Typically, this is seen in severe acute liver failure,15 with vasomotor paralysis following head injury, or occasionally in acute encephalitis. When perfusion pressure falls below the lower limit required for brain function, neurons fail to maintain their ionic gradients due to energy failure, resulting in additional swelling, which further increases ICP and results in a downward spiral of reduced perfusion and further brain infarction.

Structural Causes of Stupor and Coma |
93 |
Decreased perfusion pressure can also occur when systemic blood pressure drops, such as when assuming a standing position. Some patients with increased ICP develop brief bilateral visual loss when they stand, called visual obscurations, presumably due to failure to autoregulate the posterior cerebral blood flow. Failure of perfusion pressure can also occur focally (i.e., in a patient with an otherwise asymptomatic carotid occlusion who develops symptoms in the ipsilateral carotid distribution on standing because of the resulting small drop in blood pressure). If the patient has bilateral chronic carotid occlusions, transient loss of consciousness may result.16
Patients with elevated ICP from mass lesions often suffer sudden rises in ICP precipitated by changes in posture, coughing, sneezing, or straining, or even during tracheal suctioning (plateau waves).17 The sudden rises in ICP can reduce cerebral perfusion and produce a variety of neurologic symptoms including confusion, stupor, and coma18 (Table 3–2). In general, the symptoms last only a few minutes and then resolve, leading some observers to confuse these with seizures.
Finally, the loss of compliance of the intracranial system to further increases in volume and the rate of change in ICP plays an important
role in the response of the brain to increased ICP. Compliance is the change in pressure caused by an increase in volume. In a normal brain, increases in brain volume (e.g., due to a small intracerebral hemorrhage) can be compensated by displacement of an equal volume of CSF from the compartment. However, when a mass has increased in size to the point where there is little remaining CSF in the compartment, even a small further increase in volume can produce a large increase in compartmental pressure. This loss of compliance in cases where diffuse brain edema has caused a critical increase in ICP can lead to the development of plateau waves. These are large, sustained increases in ICP, which may approach the mean
arterial blood pressure, and which occur at intervals as often as every 15 to 30 minutes.19,20
They are thought to be due to episodic arterial vasodilation, which is due to systemic vasomotor rhythms, but a sudden increase in vascular volume in a compartment with no compliance, even if very small, can dramatically increase ICP.21 These sudden increases in ICP can thus cause a wide range of neurologic paroxysmal symptoms (see Table 3–2). When pressure in neighboring compartments is lower,
this imbalance can cause herniation (see below).22
Table 3–2 Paroxysmal Symptoms That May Result From a Sudden Increase in Intracranial Pressure
Impairment of consciousness |
Opisthotonus, trismus |
Trancelike state |
Rigidity and tonic extension/flexion |
Unreality/warmth |
of the arms and legs |
Confusion, disorientation |
Bilateral extensor plantar responses |
Restlessness, agitation |
Sluggish/absent deep tendon reflexes |
Disorganized motor activity, carphologia |
Generalized muscular weakness |
Sense of suffocation, air hunger |
Facial twitching |
Cardiovascular/respiratory disturbances |
Clonic movements of the arms and legs |
Headache |
Facial/limb paresthesias |
Pain in the neck and shoulders |
Rise in temperature |
Nasal itch |
Nausea, vomiting |
Blurring of vision, amaurosis |
Facial flushing |
Mydriasis, pupillary areflexia |
Pallor, cyanosis |
Nystagmus |
Sweating |
Oculomotor/abducens paresis |
Shivering and ‘‘goose flesh’’ |
Conjugate deviation of the eyes |
Thirst |
External ophthalmoplegia |
Salivation |
Dysphagia, dysarthria |
Yawning, hiccoughing |
Nuchal rigidity |
Urinary and fecal urgency/incontinence |
Retroflexion of the neck |
|
Adapted from Ingvar.18
94 Plum and Posner’s Diagnosis of Stupor and Coma
Conversely, when a patient shows early signs of herniation, it is often possible to reverse the situation by restoring a small margin of compliance to the compartment containing the mass lesion. Hyperventilation causes a fall in arterial pCO2, resulting in arterial and venous vasoconstriction. The small reduction in intracranial blood volume may reverse the herniation syndrome dramatically in just a few minutes.
The Role of Vascular Factors and Cerebral Edema in Mass Lesions
As indicated above, an important mechanism by which compressive lesions may cause symp-
toms is by inducing local tissue ischemia. Even in the absence of a diffuse impairment of cerebral blood flow, local increases in pressure and tissue distortion in the vicinity of a mass lesion may stretch small arteries and reduce their caliber to the point where they are no longer able to supply sufficient blood to their targets.
Many mass lesions, including tumors, inflammatory lesions, and the capsules of subdural hematomas, are able to induce the growth of new blood vessels (angiogenesis).23 These blood vessels do not have the features that characterize normal cerebral capillaries (i.e., lack of fenestrations and tight junctions between endothelial cells) that are the basis for the blood-brain barrier. Thus, the vessels leak; the leakage of
A C
 Astrocyte foot
Astrocyte foot
 Tight junction
Tight junction
Capillary endothelial cells
B
Astrocyte foot
Opened tight junctions and 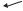 escaping plasma
escaping plasma 
Vesicular transport across endothelial cells
Edematous astrocyte
Edematous neuron
Edematous capillary endothelial cells
Figure 3–1. A schematic drawing illustrating cytotoxic versus vasogenic edema. (A) Under normal circumstances, the brain is protected from the circulation by a blood-brain barrier, consisting of tight junctions between cerebral capillary endothelial cells that do not permit small molecules to penetrate the brain, as well as a basal lamina surrounded by astrocytic end-feet. (B) When the blood-brain barrier is breached (e.g., by neovascularization in a tumor or the membranes of subdural hematoma), fluid transudates from fenestrated blood vessels into the brain. This results in an increase in fluid in the extracellular compartment, vasogenic edema. Vasogenic edema can usually be reduced by corticosteroids, which decrease capillary permeability. (C) When neurons are injured, they can no longer maintain ion gradients. The increased intracellular sodium causes a shift of fluid from the extracellular to the intracellular compartment, resulting in cytotoxic edema. Cytotoxic edema is not affected by corticosteroids. (From Fishman, RA. Brain edema. N Engl J Med 293 (14):706–11, 1975. By permission of Massachusetts Medical Society.)
contrast dyes during CT or MRI scanning provides the basis for contrast enhancement of a lesion that lacks a blood-brain barrier. The vascular leak results in the extravasation of fluid
into the extracellular space and vasogenic edema24,25 (see Figure 3–1B). This edema further
displaces surrounding tissues that are pushed progressively farther from the source of their own feeding arteries. Because the large arteries are tethered to the circle of Willis and small ones are tethered to the pial vascular system, they may not be able to be displaced as freely as the brain tissue they supply. Hence, the distensibility of the blood supply becomes the limiting factor to tissue perfusion and, in many cases, tissue survival.
Ischemia and consequent energy failure cause loss of the electrolyte gradient across the neuronal membranes. Neurons depolarize but are no longer able to repolarize and so fail. As neurons take on more sodium, they swell (cytotoxic edema), thus further increasing the mass effect on adjacent sites (see Figure 3–1C). Increased intracellular calcium meanwhile results in the activation of apoptotic programs for neuronal cell death. This vicious cycle of swelling produces ischemia of adjacent tissue, which in turn causes further tissue swelling. Cytotoxic edema may cause a patient with a chronic and
slowly growing mass lesion to decompensate quite suddenly,24,25 with rapid onset of brain
failure and coma when the lesion reaches a critical limit.
HERNIATION SYNDROMES: INTRACRANIAL SHIFTS IN THE PATHOGENESIS OF COMA
The Monro-Kellie doctrine hypothesizes that because the contents of the skull are not compressible and are contained within an unyielding case of bone, the sum of the volume of the brain, CSF, and intracranial blood is constant at all times.26 A corollary is that these same restrictions apply to each compartment (right vs. left supratentorial space, infratentorial space, spinal subarachnoid space). In a normal brain, increases in the size of a growing mass lesion can be compensated for by the displacement of an equal volume of CSF from the compartment. The displacement of CSF, and in some cases blood volume, by the mass lesion raises ICP. As the mass grows, there is less CSF to be
Structural Causes of Stupor and Coma |
95 |
displaced, and hence the compliance of the intracranial contents decreases as the size of the compressive lesion increases. When a mass has increased in size to the point where there is little remaining CSF in the compartment, even a small further increase in volume can produce a large increase in compartmental pressure. When pressure in neighboring compartments is lower, this imbalance causes herniation. Thus, intracranial shifts are of key concern in the diagnosis of coma due to supratentorial mass lesions (Figure 3–2).
The pathogenesis of signs and symptoms of an expanding mass lesion that causes coma is rarely a function of the increase in ICP itself, but usually results from imbalances of pressure between different compartments leading to tissue herniation.
To understand herniation syndromes, it is first necessary to review briefly the structure of the intracranial compartments between which herniations occur.
Anatomy of the Intracranial
Compartments
The cranial sutures of babies close at about 18 months, encasing the intracranial contents in a nondistensible box of finite volume. The intracranial contents include the brain tissue (approximately 87%, of which 77% is water), CSF (approximately 9%), blood vessels (approximately 4%), and the meninges (dura, arachnoid, and pia that occupy a negligible volume). The dural septa that divide the intracranial space into compartments play a key role in the herniation syndromes caused by supratentorial mass lesions.
The falx cerebri (Figures 3–2 and 3–3) separates the two cerebral hemispheres by a dense dural leaf that is tethered to the superior sagittal sinus along the midline of the cranial vault. The falx contains the inferior sagittal sinus along its free edge. The free edge of the falx normally rests just above the corpus callosum. One result is that severe head injury can cause a contusion of the corpus callosum by violent upward displacement of the brain against the free edge of the falx.33 The pericallosal branches of the anterior cerebral artery also run in close proximity to the free edge of the falx. Hence, displacement of the cingulate gyrus under the falx by a hemispheric mass may compress the pericallosal artery and result in ischemia or infarction of