

Книги по МРТ КТ на английском языке / PLUM AND POSNER S DIAGNOSIS OF STUPOR AND COM-1
.pdf106 Plum and Posner’s Diagnosis of Stupor and Coma
usually ptosis (if indeed the patient opens the eyes at all).
The lapse into coma may take place over just a few minutes, as in the patient above who was uncooperative with the x-ray technician and 10 minutes later was found by the neurologist to be deeply comatose. Hemiparesis may be ipsilateral to the herniation (if the midbrain is compressed against the opposite tentorial edge) or may be contralateral (if the paresis is due to the lesion damaging the descending corti-
cospinal tract or to a herniating temporal lobe compressing the ipsilateral cerebral peduncle). Breathing is typically normal, or the patient may lapse into a Cheyne-Stokes pattern of respiration (Figure 3–10).
MIDBRAIN-UPPER PONTINE STAGE
If treatment is delayed or unsuccessful, signs of midbrain damage appear and progress caudally, as in central herniation (see below). Both pupils
a. Respiratory |
or |
pattern |
|
Regular sustained hyperventilation |
Rarely, Cheyne-Stokes |
b. Pupillary |
|
|
size and |
|
|
reactions |
ipsilateral pupil widely |
Does not constrict |
|
||
|
dilated |
|
c. Oculocephalic and oculovestibular responses
DOLL’S HEAD MANEUVER |
ICE WATER CALORICS |
d.Motor responses at rest and to stimulation
Ipsilateral eye doesn’t move medially, but contralateral eye retains full lateral movement
Decorticate or decerebrate responses
Figure 3–10. Signs of uncal herniation, late third nerve stage.
may fix at midposition, and neither eye elevates, depresses, or turns medially with oculocephalic or caloric vestibular testing. Either decorticate or decerebrate posturing may be seen.
Clinical Findings in Central
Herniation Syndrome
DIENCEPHALIC STAGE
The first evidence that a supratentorial mass is beginning to impair the diencephalon is usually a change in alertness and behavior. Initially, subjects might find it difficult to concentrate and may be unable to retain the orderly details of recent events. As the compression of the diencephalon progresses, the patient lapses into torpid drowsiness, and finally stupor and coma.
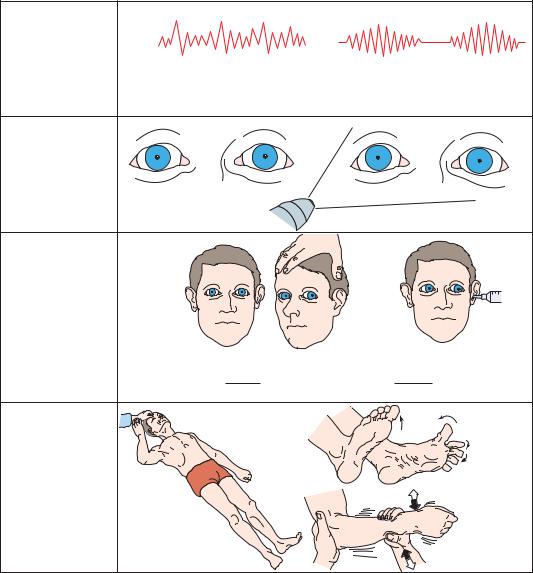
Respiration in the early diencephalic stage of central herniation is commonly interrupted by sighs, yawns, and occasional pauses (Figure 3–11). As the sleepiness deepens, many patients lapse into the periodic breathing of Cheyne-Stokes respiration. The pupils are typically small (1 to 3 mm), and it may be difficult to identify their reaction to light without a bright light source or a magnifying glass. However, the pupils typically dilate briskly in response to a pinch of the skin over the neck (ciliospinal reflex).58 The eyes are typically conjugate or slightly divergent if the patient is not awake, and there may be roving eye movements, with slow to-and-fro rolling conjugate displacement. Oculocephalic testing typically demonstrates brisk, normal responses. There is typically a diffuse, waxy increase in motor tone (paratonia or gegenhalten), and the toe signs may become bilaterally extensor.
The appearance of a patient in the early diencephalic stage of central herniation is quite similar to that in metabolic encephalopathy. This is a key problem, because one would like to identify patients in the earliest phase of central herniation to institute specific therapy, and yet these patients look most like patients who have no structural cause of coma. For this reason, every patient with the clinical appearance of metabolic encephalopathy requires careful serial examinations until a structural lesion can be ruled out with an imaging study and a metabolic cause of coma can be identified and corrected.
During the late diencephalic stage (Figure 3–12), the clinical appearance of the patient
Structural Causes of Stupor and Coma |
107 |
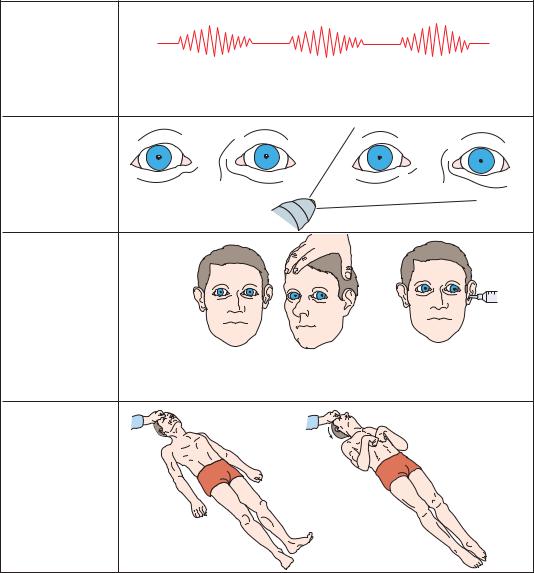
becomes more distinctive. The patient becomes gradually more difficult to arouse, and eventually localizing motor responses to pain may disappear entirely or decorticate responses may appear. Initially, the upper extremity flexor and lower extremity extensor posturing tends to appear on the side contralateral to the lesion, and only in response to noxious stimuli. Later, the response may become bilateral, and eventually the contralateral and then ipsilateral side may progress tofullextensor (decerebrate) posturing.
The mechanism for brain impairment during the diencephalic stage of central herniation is not clear. Careful quantitative studies show that the depressed level of consciousness correlates with either lateral or vertical displacement of the pineal gland, which lies along the midline at the rostral extreme of the dorsal midbrain.59,60 The diencephalic impairment may be due to the stretching of small penetrating vessels tethered to the posterior cerebral and communicating arteries that supply the caudal thalamus and hypothalamus. There is little evidence that either increases in ICP or changes in cerebral blood flow can account for these findings. On the other hand, if patients with diencephalic signs of the central herniation syndrome worsen, they tend to pass rapidly to the stage of midbrain damage, suggesting that the same pathologic process has merely extended to the next more caudal level.
The clinical importance, therefore, of the diencephalic stage of central herniation is that it warns of a potentially reversible lesion that is about to encroach on the brainstem and create irreversible damage. If the supratentorial process can be alleviated before the signs of midbrain injury emerge, chances for a complete neurologic recovery are good. Once signs of lower diencephalic and midbrain dysfunction appear, it becomes increasingly likely that they will reflect infarction rather than compression and reversible ischemia, and the outlook for neurologic recovery rapidly becomes much poorer.
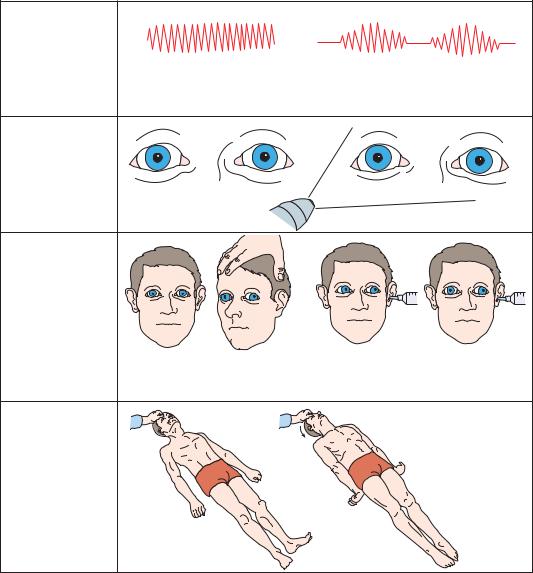
As herniation progresses to the midbrain stage (Figure 3–13), signs of oculomotor failure appear. The pupils become irregular, then fixed at midposition. Oculocephalic movements become more difficult to elicit, and it may be necessary to examine cold water caloric responses to determine their full extent. Typically, there is limited and slower, and finally no medial movement of the eye contralateral to the cold water stimulus, and bilateral warm or cold water irrigation
108 Plum and Posner’s Diagnosis of Stupor and Coma
a. Respiratory |
|
|
pattern |
Eupneic, with deep sighs |
Cheyne-Stokes |
|
||
|
or yawns |
|
b. Pupillary |
|
|
size and |
|
|
reactions |
Small pupils |
Small range of contraction |
|
c. Oculocephalic |
|
and |
|
oculovestibular |
|
responses |
|
DOLL’S HEAD MANEUVER |
ICE WATER CALORICS |
Full conjugate lateral, |
Full conjugate lateral, |
opposite to direction |
ipsilateral to ear injected |
of turning |
|
|
Bilateral |
|
Babinski’s |
d. Motor |
|
responses |
|
at rest |
|
and to |
|
stimulation |
Paratonic |
|
resistance |
Appropriate motor |
|
response to noxious |
|
orbital roof |
|
pressure |
|
Figure 3–11. Signs of central transtentorial herniation or lateral displacement of the diencephalon, early diencephalic stage.
confirms lack of vertical eye movements. Motor responses are difficult to obtain or result in extensor posturing. In some cases, extensor posturing appears spontaneously, or in response to internal stimuli. Motor tone and tendon reflexes may be heightened, and plantar responses are extensor.
After the midbrain stage becomes complete, it is rare for patients to recover fully. Most patients in whom the herniation can be reversed suffer chronic neurologic disability.61,62
Hence, it is critical, if intervention is anticipated, that it begin as early as possible and that it be as vigorous as possible, as the patient’s life hangs in the balance.
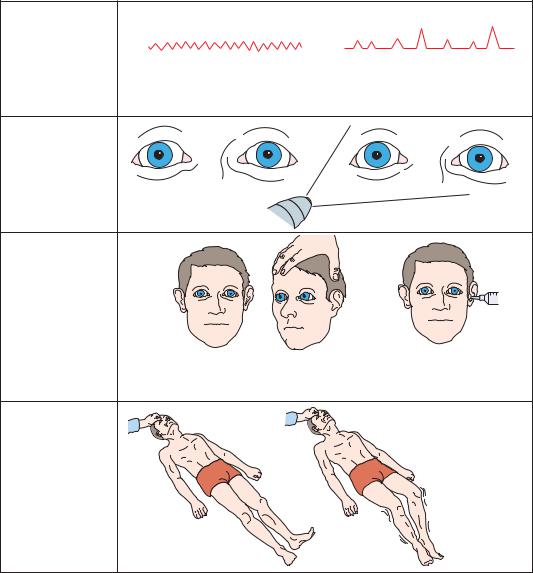
As the patient enters the pontine stage (Figure 3–14) of herniation, breathing becomes more shallow and irregular, as the upper pontine structures that modulate breathing are lost. As the damage approaches the lower pons, the lateral eye movements produced by cold water caloric stimulation are also lost. Motor tone be-
Structural Causes of Stupor and Coma |
109 |
a. Respiratory
pattern
Cheyne-Stokes
b. Pupillary |
|
|
size and |
|
|
reactions |
Small pupils |
Small range of contraction |
|
c. Oculocephalic |
|
and |
|
oculovestibular |
|
responses |
|
DOLL’S HEAD MANEUVER |
ICE WATER CALORICS |
Same as Fig 3–11, but easier |
Same as Fig. 3–11 but easier |
to obtain (absent nystagmus) |
to obtain (absent nystagmus) |
Motionless |
Legs stiffen and arms |
d. Motor |
rigidly flex |
responses |
(decorticate rigidity) |
at rest |
|
and to |
|
stimulation |
|
Figure 3–12. Signs of central transtentorial herniation, or lateral displacement of the diencephalon, late diencephalic stage.
comes flaccid, tendon reflexes may be difficult to obtain, and lower extremity posturing may become flexor.
The medullary stage is terminal. Breathing becomes irregular and slows, often assuming a gasping quality. As breathing fails, sympathetic reflexes may cause adrenalin release, and the pupils may transiently dilate. However, as cerebral hypoxic and baroreceptor reflexes also become impaired, autonomic reflexes fail and blood pressure drops to levels seen after high
spinal transection (systolic pressures of 60 to 70 mm Hg).
At this point, intervening with artificial ventilation and pressor drugs may keep the body alive, and all too often this is the reflexive response in a busy intensive care unit. It is important to recognize, however, that once herniation progresses to respiratory compromise, there is no chance of useful recovery. Therefore, it is important to discuss the situation with the family of the patient before the onset of the medullary stage,
110 Plum and Posner’s Diagnosis of Stupor and Coma
a. Respiratory |
|
|
pattern |
Sustained regular |
Rarely, Cheyne-Stokes |
|
||
|
hyperventilation |
|
|
|
|
b. Pupillary |
|
|
size and |
|
|
reaction |
Midposition often |
Fixed |
|
||
|
irregular in shape |
|
c. Oculocephalic |
|
|
and |
|
|
oculovestibular |
|
|
responses |
|
|
DOLL’S HEAD MANEUVER |
ICE WATER CALORICS |
|
Impaired, may be |
Impaired, may be |
|
dysconjugate |
dysconjugate |
|
|
Arms and legs |
|
d. Motor |
extend and pronate |
|
(decerebrate rigidity) |
||
responses |
particularly on side |
|
at rest |
opposite primary |
|
lesion |
||
and to |
||
or |
||
stimulation |
||
|
||
Usually |
|
|
motionless |
|
Figure 3–13. Signs of transtentorial herniation, midbrain-upper pons stage.
and to make it clear that mechanical ventilation in this situation merely prolongs the process of dying.
Clinical Findings in Dorsal
Midbrain Syndrome
The midbrain may be forced downward through the tentorial opening by a mass lesion impinging upon it from the dorsal surface (Figure 3–15).
The most common causes are masses in the pineal gland (pinealocytoma or germ cell line tumors) or in the posterior thalamus (tumor or hemorrhage into the pulvinar, which normally overhangs the quadrigeminal plate at the posterior opening of the tentorial notch). Pressure from this direction produces the characteristic dorsal midbrain syndrome. A similar picture may be seen during upward transtentorial herniation, which kinks the midbrain (Figure 3–8).
Structural Causes of Stupor and Coma |
111 |
a. Respiratory |
or |
|
|
pattern |
|
Eupneic, although often more |
Slow and irregular in rate |
shallow and rapid than normal |
and amplitude (ataxic) |
b. Pupillary |
|
|
size and |
|
|
reaction |
Midposition |
Fixed |
|
c. Oculocephalic |
|
|
and |
|
|
oculovestibular |
|
|
responses |
|
|
|
DOLL’S HEAD MANEUVER |
ICE WATER CALORIC |
|
No response |
No response |
|
|
No response to |
|
|
noxious orbital |
d. Motor |
|
stimulus; bilateral |
|
Babinski signs or |
|
responses |
|
|
|
occasional flexor |
|
at rest |
|
response in lower |
and to |
or |
extremities when |
stimulation |
|
feet stroked |
|
|
Motionless and flaccid
Figure 3–14. Signs of transtentorial herniation, lower pons-upper medulla stage.
Pressure on the olivary pretectal nucleus and the posterior commissure produces slightly enlarged (typically 4 to 6 mm in diameter) pupils that are fixed to light.2 There is limitation of vertical eye movements, typically manifested first by limited upgaze. In severe cases, the eyes may be fixed in a forced, downward position. If the patient is awake, there may also be a deficit of convergent eye movements and associated pupilloconstriction. The presence of retractory nystagmus, in which all of the eye
muscles contract simultaneously to pull the globe back into the orbit, is characteristic. Retraction of the eyelids may produce a staring appearance.
Deficits of arousal are present in only about
15% of patients with pineal region tumors, but these are due to early central herniation.63,64
If the cerebral aqueduct is compressed sufficiently to cause acute hydrocephalus, however, an acute increase in supratentorial pressure may ensue. This may cause an acute increase in
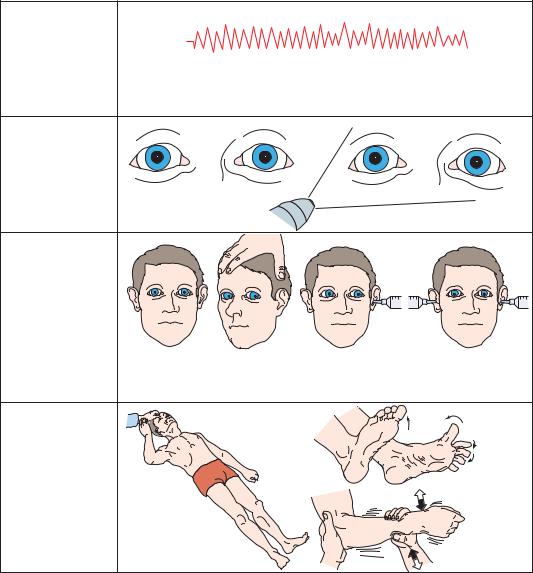
112Plum and Posner’s Diagnosis of Stupor and Coma
a.Respiratory pattern
Eupneic
b. Pupillary |
|
|
size and |
|
|
reactions |
Moderately dilated |
No reaction to light |
|
c. Oculocephalic |
|
|
and |
|
|
oculovestibular |
|
|
responses |
|
|
DOLL’S HEAD MANEUVER |
ICE WATER CALORICS |
|
Downward with full lateral movements. |
Downward with full |
Downward with no |
Early loss of upgaze and vergence |
lateral movements |
upward movement |
then downgaze. |
|
(bilateral cold water) |
|
|
Bilateral |
|
|
Babinski’s |
d. Motor |
|
|
responses |
|
|
at rest |
|
|
and to |
|
|
stimulation |
|
Paratonic |
|
|
resistance |
Appropriate motor |
|
|
response to noxious |
|
|
orbital roof |
|
|
pressure |
|
|
Figure 3–15. Signs of dorsal midbrain compression.
downward pressure on the midbrain, resulting in sudden lapse into deep coma.
Safety of Lumbar Puncture
in Comatose Patients
A common question encountered clinically is, ‘‘Under what circumstances is lumbar puncture safe in a patient with an intracranial mass lesion?’’ There is often a large pressure gradi-
ent between the supratentorial compartment and the lumbar sac,65 and lowering the lumbar pressure by removing CSF may increase the gradient. The actual frequency of cases in which this hypothetical risk causes transtentorial herniation is difficult to ascertain. Most available studies date to the pre-CT era, as clinicians perform a lumbar puncture only rarely after the presence of a supratentorial mass lesion of any size is identified. Several older studies examining series of patients with brain
tumors who underwent lumbar puncture found complication rates in the range of 1% to 2% in
patients with documented increased CSF pressure or papilledema.66,67 On the other hand,
of patients referred to a neurosurgical service because of complications following lumbar puncture, Duffy reported that 22 had focal neurologic signs before the lumbar puncture, but only one-half had increased CSF pressure and one-third had papilledema.56 The experience of the authors supports the view that although lumbar puncture rarely precipitates transtentorial (or foramen magnum) herniation, even in patients who may be predisposed by an existing supratentorial mass lesion, neither the physical examination nor the evaluation of CSF pressure at lumbar puncture is sufficient to predict which patients will suffer complications. Hence, before any patient undergoes lumbar puncture, it is wise to obtain a CT (or MRI) scan of the cranial contents. If a patient has no evidence of compartmental shift on the study, it is quite safe to obtain a lumbar puncture. On the other hand, if it is impossible to obtain an imaging study in a timely fashion and the neurologic examination shows no papilledema or focal signs, the risk of lumbar puncture is quite low (probably less than 1%). Under such circumstances, risk-benefit analysis may well favor proceeding with lumbar puncture if the study is needed to make potentially lifesaving decisions about clinical care.
False Localizing Signs in the
Diagnosis of Structural Coma
It is usually relatively easy for a skilled examiner to differentiate supratentorial from infratentorial signs, and the cranial nerve findings due to herniation syndromes are characteristic. However, there are a number of specific situations in which the neurologic signs may falsely cause the examiner to consider an infratentorial process or to mistake an infratentorial process for one that is supratentorial.
The most common false localizing sign is abducens palsy. This may be caused by increased ICP, or it may occur after lumbar puncture. In the latter case, the reduced CSF volume can cause the brain to lose its usual buoyant suspension in the CSF. The sagging of the brain in an upright posture is thought to cause traction on the abducens nerve. More rarely other cra-
Structural Causes of Stupor and Coma |
113 |
nial nerves, including the trochlear, oculomotor, or trigeminal nerves, may be similarly affected.
Differentiation of supratentorial from infratentorial causes of ataxia has presented a diagnostic dilemma since the earliest days of neurology.68 In the days before imaging, despite highly developed clinical skills, it was not unusual for a neurosurgeon to explore the posterior fossa, find nothing, and then turn the patient over and remove a frontal tumor. The gait disorder that is associated with bilateral medial frontal compression or hydrocephalus can be replicated on occasion by cerebellar lesions. Similarly, unilateral ataxia of finger-nose-finger testing, which appears to be cerebellar in origin, may occasionally be seen with parietal lobe lesions.69
Another source of confusion is the differentiation of upper (supranuclear) versus lower motor neuron cranial nerve palsies. Although rare, acute supratentorial lesions can on occasion cause lower cranial nerve palsies (asymmetric palate, tongue weakness on one side). Bilateralsupratentoriallesionscanproduce dysarthria, dysphagia, and bilateral facial weakness (pseudobulbar palsy, also called the opercular or Foix-Chavany-Marie syndrome70). Conversely, the well-known upper motor neuron facial palsy (weakness of the lower part of the face) can be seen with some posterior fossa lesions. The distinction between upper versus lower motor neuron cranial nerve weakness can often be made on the basis of reflex versus voluntary movement. For example, a patient with supranuclear bulbar weakness will often show intact, or even hyperactive, corneal or gag reflexes. A patient with an upper motor neuron facial palsy will typically show a much more symmetric smile on responding to a joke than when asked to smile voluntarily.
Fortunately, these classic problems with localization rarely intrude on interpretation of the examination of a patient with an impaired level of consciousness, as the signs associated with herniation typically develop relatively rapidly as the patient loses consciousness. If the patient displays false localizing signs while awake, the progression of new signs that occur during the herniation process generally clarifies the matter. Nearly all patients with impaired consciousness and focal brainstem signs should be treated as structural coma and receive immediate imaging studies, so that any
114 Plum and Posner’s Diagnosis of Stupor and Coma
confusion about the source of the findings should be short-lived.
DESTRUCTIVE LESIONS
AS A CAUSE OF COMA
Destructive lesions of the ascending arousal system or its forebrain targets are paradoxically less of an immediate diagnostic concern for the examining neurologist. Unlike compressive lesions, which can often be reversed by removing a mass, destructive lesions typically cannot be reversed. Although it is important to recognize the hallmarks of a destructive, as opposed to a compressive, lesion, the real value comes in distinguishing patients who may benefit from immediate therapeutic intervention from those who need mainly supportive care.
DIFFUSE, BILATERAL CORTICAL DESTRUCTION
Diffuse, bilateral destruction of the cerebral cortex or its underlying white matter can occur as a result of deprivation of metabolic substrate (i.e., oxygen, glucose, or the blood that carries them) or as a consequence of certain metabolic or infectious diseases. This condition is often the consequence of prolonged cardiac arrest in a patient who is eventually resuscitated, but it may also occur in patients who have diffuse hypoxia due to pulmonary failure or occasionally in patients with severe and prolonged hypoglycemia. The lack of metabolic substrate causes neurons in layers III and V of the cerebral cortex, and in the CA1 and CA3 fields of the hippocampal formation, to be damaged,71,72 presumably as a result of excitatory amino acid toxicity (see Figure 1–10). During periods of metabolic deprivation, there is rundown of the ion gradients that support normal membrane polarization, resulting in depolarization of neurons and release of their neurotransmitters. Excess excitatory transmitter, particularly acting on N-methyl-d-aspartate (NMDA) receptors that allow the intracellular shift of calcium ions, results in activation of genetic neuronal death programs and the elimination of neurons that receive intense excitatory amino acid inputs.73 Because excitatory amino acids are used extensively for corticocortical communication, the neurons that are at greatest risk are those that
receive those connections, which in turn are the ones that are most responsible for cortical output as well. The remaining neurons are essentially cut off from one another and from their outputs, and thus are unable to provide meaningful behavioral response.
Patients who have suffered from a period of hypoxia of somewhat lesser degree may appear to recover after brain oxygenation is restored. However, over the following week or so there may be a progressive degeneration of the subcortical white matter, essentially isolating the cortex from its major inputs and outputs.74 This condition is seen most commonly after carbon monoxide poisoning (see page 30), but may occur after other sublethal episodes of hypoxia. The mechanism of white matter injury is not known, although it may be related to similar white matter injury that is seen in Leigh’s disease, mitochondrial encephalopathy with ‘‘strokes,’’ and other disorders of cerebral intermediary metabolism that leave the brain with inadequate but sublethal impairment of oxidative energy metabolism.75
Other metabolic leukoencephalopathies, such as metachromatic leukodystrophy and Canavan’s disease, rarely occur in adults but are considerations when assessing infants or very young children. Adrenoleukodystrophy may cause mainly posterior hemispheric white matter disease, but rarely affects the level of consciousness until very late in the disease.
Infectious causes of dysfunction of the cerebral cortex or subjacent white matter include prion infections (Creutzfeldt-Jakob disease, Gerstmann-Stra¨ussler syndrome, etc.) and progressive multifocal leukoencephalopathy. These disorders progress over a period of weeks to months, and so rarely present a diagnostic dilemma by the time global consciousness is impaired. Subacute sclerosing panencephalitis, due to slow viral infection with the measles virus, can also cause this picture, but it is rarely seen in populations in which measles vaccination is practiced.
DESTRUCTIVE DISEASE OF THE DIENCEPHALON
Bilateral destructive lesions of the diencephalon are a rare cause of coma, in part because the diencephalon receives its blood supply directly from feeding vessels that take off from the major
arteries of the circle of Willis. Hence, although vascular disease may affect the diencephalon when any one major arterial source is compromised, it is typically unilateral and does not impair consciousness. An exception occurs when there is occlusion of the tip of the basilar artery, which supplies the posterior cerebral and communicating arteries bilaterally. The posterior thalamic penetrating arteries take their origin from these posterior components of the circle of Willis, and as a consequence there may be bilateral posterior thalamic infarction with a single site of vascular occlusion.76 However, nearly all cases in which there is impairment of consciousness also have some midbrain ischemia as well (see vascular causes of coma in Chapter 4, page 152).
Occasional inflammatory and infectious disorders may have a predilection for the diencephalon. Fatal familial insomnia, a prion disorder, is reported to affect the thalamus selectively, and this has been proposed as a cause of the sleep disorder (although this produces hyperwakefulness, not coma).77 Behc¸et’s disease may cause sterile abscess formation in the diencephalon, which may depress the level of consciousness.78
Autoimmune disorders may also affect the diencephalon. In patients with anti-Ma antitumor antibodies, there are often diencephalic lesions as well as excessive sleepiness and sometimes other symptoms of narcolepsy, such as cataplexy.79 It is now recognized that in most patients with narcolepsy, there is a progressive loss of neurons in the lateral hypothalamus that express the neurotransmitter orexin, also called hypocretin.80,81 This selective loss of orexin neurons is believed to be autoimmune in origin, although this remains to be demonstrated definitively.82 Loss of orexin neurons results in excessive sleepiness, but should not cause impairment of consciousness while awake.
Rarely, primary brain tumors may arise in the diencephalon. These may be either astrocytomas or primary central nervous system lymphomas, and they can cause impairment of consciousness as an early sign. Suprasellar tumors such as craniopharyngioma or suprasellar germinoma, or suprasellar extension of a large pituitary adenoma, can compress the diencephalon, but does not usually cause destruction unless attempts at surgical excision cause local vasospasm.83
Structural Causes of Stupor and Coma |
115 |
DESTRUCTIVE LESIONS
OF THE BRAINSTEM
In destructive disorders of the brainstem, acute loss of consciousness is typically accompanied by a distinctive pattern of pupillary, oculomotor, motor, and respiratory signs that indicate the level of the brainstem that has been damaged. Unlike rostrocaudal deterioration, however, in which all functions of the brainstem above the level are lost, tegmental lesions of the brainstem often are accompanied by more limited findings that pinpoint the level of the lesion.
Destructive lesions at the level of the midbrain tegmentum typically destroy the oculomotor nuclei bilaterally, resulting in fixed midposition pupils and paresis of adduction, elevation, and depression of the eyes. At the same time, the abduction of the eyes with oculocephalic maneuvers is preserved. If the cerebral peduncles are also damaged, as with a basilar artery occlusion, there is bilateral flaccid paralysis.
A destructive lesion of the rostral pontine tegmentum spares the oculomotor nuclei, so that the pupils remain reactive to light. If the lateral pontine tegmentum is involved, the descending sympathetic and ascending pupillodilator pathways are both damaged, resulting in tiny pupils whose reaction to light may be discernible only by using a magnifying glass. Damage to the medial longitudinal fasciculus causes loss of adduction, elevation, and depression in response to vestibular stimulation, but abduction is preserved, as are behaviorally directed vertical and vergence eye movements. If the lesion extends somewhat caudally into the midpons, there may be gaze paresis toward the side of the lesion or slow vertical eye movements, called ocular bobbing, or its variants (Table 2–3). When the lesion involves the base of the pons, there may be bilateral flaccid paralysis. However, this is not necessarily seen if the lesion is confined to the pontine tegmentum. Facial or trigeminal lower motor neuron paralysis can also be seen if the lesion extends into the more caudal pons. Involvement of the pons may also produce apneustic or ataxic breathing.
On the other hand, destructive lesions that are confined to the lower pons or medulla do not cause loss of consciousness.84 Such patients may, however, have sufficient damage to the