

Книги по МРТ КТ на английском языке / PLUM AND POSNER S DIAGNOSIS OF STUPOR AND COM-1
.pdf156 Plum and Posner’s Diagnosis of Stupor and Coma
Table 4–13 Symptoms and Signs in 78 Reported Cases of Patients With Documented Central Nervous System Granulomatous Angitis
|
|
Total No. Recorded |
|
No. at |
During the Course |
Symptom/Sign |
Onset |
of the Disease |
|
|
|
Mental changes |
45 |
61 |
Headache |
42 |
42 |
Coma |
0 |
42 |
Focal weakness |
12 |
33 |
Seizure |
9 |
18 |
Fever |
16 |
16 |
Ataxia |
7 |
11 |
Aphasia |
4 |
10 |
Visual changes (diplopia, amaurosis, |
|
|
and blurring) |
7 |
9 |
Tetraparesis |
0 |
9 |
Flaccid or spastic paraparesis |
7 |
8 |
(with back pain, sensory level, |
|
|
and urinary incontinence) |
|
|
From Younger et al.,217 with permission.
anticoagulation and thrombolytic therapy are believed to be effective210,215; some thrombi
recanalize spontaneously.
Vasculitis
Vasculitis affecting the brain either can occur as part of a systemic disorder216 (e.g., polyarteritis nodosa, Behc¸et’s syndrome) or can be restricted to the nervous system (e.g., CNS granulomatous angiitis).217 The disorder can affect large or small vessels. Vasculitis causes impairment of consciousness by ischemia or infarction that either affects the hemispheres diffusely or the brainstem arousal systems. The diagnosis can be suspected in a patient with headache, fluctuating consciousness, and focal neurologic signs (Table 4–13). Granulomatous angiitis, the most common CNS vasculitis, is discussed here. Other CNS vasculopathies are discussed in Chapter 5 (see page 273).
The CSF may contain an increased number of lymphocytes or may be normal. The CT scan may likewise be normal, but MRI usually demonstrates areas of ischemia or infarction. Magnetic resonance angiography may demonstrate multifocal narrowing of small blood vessels or may be normal. High-resolution arteriography
is more likely to demonstrate small vessel abnormalities. A definitive diagnosis can be made only by biopsy. Even then, because of sampling error, biopsy may not establish the diagnosis. The treatment depends on the cause of vasculitis; most of the disorders are immune mediated and are treated by immunosuppression, usually with corticosteroids and cyclophosphamide.218
INFECTIONS AND
INFLAMMATORY CAUSES
OF SUPRATENTORIAL
DESTRUCTIVE LESIONS
Viral Encephalitis
Although bacteria, fungi, and parasites can all invade the brain (encephalitis) with or without involvement of the meninges (meningoencephalitis), they tend to form localized infections. Viral encephalitis, by distinction, is often widespread and bilateral, and hence coma is a common feature. The organisms destroy tissue both by direct invasion and as a result of the immune response to the infectious agent. They may further impair neurologic function as toxins

Specific Causes of Structural Coma |
157 |
Table 4–14 Findings in 113 Patients With Herpes Simplex Encephalitis
|
No. (%) of |
|
No. (%) of |
|
Patients |
|
Patients |
|
|
|
|
Historic Findings |
|
Clinical Findings |
|
Alteration of consciousness |
109/112 (97) |
at Presentation |
|
Cerebrospinal fluid pleocytosis |
107/110 (97) |
Fever |
101/110 (92) |
Fever |
101/112 (90) |
Personality change |
69/81 (85) |
Headache |
89/110 (81) |
Dysphasia |
58/76 (76) |
Personality change |
62/87 (71) |
Autonomic dysfunction |
53/88 (60) |
Seizures |
73/109 (67) |
Ataxia |
22/55 (40) |
Vomiting |
51/111 (46) |
Hemiparesis |
41/107 (38) |
Hemiparesis |
33/100 (33) |
Seizures |
43/112 (38) |
Memory loss |
14/59 (24) |
Focal |
28 |
Cranial nerve defects |
34/105 (32) |
Generalized |
10 |
|
|
Both |
5 |
|
|
Visual field loss |
8/58 (14) |
|
|
Papilledema |
16/111(14) |
From Whitley et al.,220 with permission.
produced by the organisms or cytokines or prostaglandins in response to the presence of the organisms may interfere with neuronal function.
Although many different organisms can cause encephalitis, including a number of mosquitoborne viruses with regional variations in prevalence (eastern and western equine, St. Louis, Japanese, and West Nile viruses), by far the most common and serious cause of sporadic encephalitis is herpes simplex type I.219 This disorder accounts for 10% to 20% of all viral infections of the CNS. Patients characteristically have fever, headache, and alteration of consciousness that culminate in coma (Table 4– 14). Personality changes, memory impairment, or seizures focus attention on the medial temporal, frontal, and insular areas, where the infection usually begins and is most severe.
Routine examination of CSF is not very helpful. There is usually a pleocytosis with a white count of as many as 100 cells and a protein concentration averaging 100 mg/dL. Red cells may or may not be present. As many as 10% of patients may have a normal CSF examination when initially seen. However, polymerase chain reaction (PCR) detection of herpes simplex virus in CSF is diagnostic. The EEG may be helpful if it shows slowing or epileptiform activity arising from the temporal lobe. CT and MRI are very helpful, showing edema and then destruction predominantly in the temporal and frontal lobes, and often in the
insular cortex (Figure 4–11). The destruction can initially be unilateral but usually rapidly becomes bilateral. The differential diagnosis includes other forms of encephalitis including bacteria and viruses, and even low-grade astrocytomas of the medial temporal lobe, which may present with seizures and a subtle low density lesion.
It is very important to begin treatment as early as possible with an antiviral agent such as acyclovir at 10 mg/kg every 8 hours for 10 to 14 days.221 Most patients who are treated promptly make a full recovery, although an occasional patient is left with severe memory loss.
Acute Disseminated
Encephalomyelitis
Acute disseminated encephalomyelitis (ADEM) is an allergic, presumably autoimmune, encephalitis that is seen during or after an infectious illness, but which may also be caused by vaccination. Spontaneous sporadic cases are
believed to result from a subclinical infectious illness.222,223 Patients develop multifocal neu-
rologic symptoms, usually over a period of several days, about 1 to 2 weeks after a febrile illness. Neurologic signs may include a wide variety of sensory and motor complaints, as they do in patients with multiple sclerosis, but a
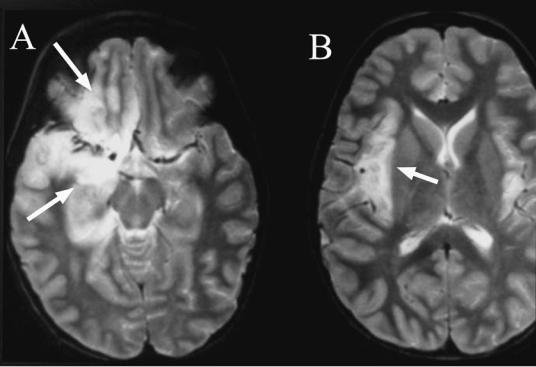
158 Plum and Posner’s Diagnosis of Stupor and Coma
Figure 4–11. A pair of magnetic resonance images from the brain of a patient with herpes simplex 1 encephalitis. Note the preferential involvement of the medial temporal lobe and orbitofrontal cortex (arrows in A) and insular cortex (arrow in B). There is milder involvement of the contralateral side.
key differentiating point is that a much larger percentage of patients with ADEM present with behavioral disturbances, whereas this is rare early in multiple sclerosis. Occasionally patients with ADEM may become stuporous or comatose (see Patient 4–4), findings that are also rare in early multiple sclerosis. CT or MRI scan shows multifocal enhancing lesions in the white matter, but these may appear late in the illness (see Patient 4–4). Although the pathology is distinct from multiple sclerosis, showing mainly perivascular infiltration and demyelination, the appearance of the lesions on MRI scan is essentially identical in the two illnesses. CSF may show 100 or more white blood cells and an elevation of protein, but may show no changes at all; oligoclonal bands are often absent.
In most cases, it is difficult to distinguish ADEM from first onset of multiple sclerosis. The likelihood of ADEM is increased if the patient has recently had a febrile illness, if the illness is dominated by behavioral or cogni-
tive problems or impairment of consciousness, or if there are large plaques in the hemispheric white matter. However, the proof of the diagnosis is established by the course of the illness. Although ADEM can fluctuate, and new symptoms and plaques can continue to appear for up to several weeks, it is essentially a monotonic illness, whereas new lesions appearing after 1 or more months generally portend the diagnosis of multiple sclerosis. Overall, in various series approximately one-third of patients initially diagnosed with ADEM go on to develop multiple sclerosis.
Treatment of ADEM also differs from multiple sclerosis. Although there has been no randomized, controlled series, in our experience patients often improve dramatically with oral prednisone, 40 to 60 mg daily. The dose is then tapered to the lowest maintenance level that does not allow recrudescence of symptoms. However, the patient may require oral steroid treatment for months, or even a year or two.

Patient 4–4
A 42-year-old secretary had pharyngitis, fever, nausea, and vomiting, followed 3 days later by confusion and progressive leg weakness. She came to the emergency department, where she was found to have a stiff neck, left abducens palsy, and moderate leg weakness, with a sensory level at around T8 to pin. She rapidly became stuporous, then comatose, with flaccid quadriplegia.
Spinal fluid showed 81 white blood cells/mm3, with 87% lymphocytes, protein 66 mg/dL, and glucose 66 mg/dL. An MRI scan of the brain and the spinal cord, including contrast, at the time of onset of impaired consciousness and then again 2 days later did not show any abnormalities. She required intubation and mechanical ventilation. A repeat MRI scan on day 8 demonstrated patchy, poorly marginated areas of T2 signal hyperintensity in the white matter of both cerebral hemispheres, the brainstem, and the cerebellum, consistent with ADEM. She was treated with corticosteroids and over a period of 3 months, recovered, finished rehabilitation, and was able to resume her career and playing tennis.
CONCUSSION AND OTHER TRAUMATIC BRAIN INJURIES
Traumatic brain injury, a common cause of coma, is usually easily established because there is a history or external signs of head injury at the time of presentation. Nevertheless, because so many traumatic events occur in individuals who are already impaired by drug ingestion or comorbid illnesses (e.g., hypoglycemia in a diabetic), other causes of loss of consciousness must always be considered. The nature of the traumatic intracranial process that produces impairment of consciousness requires rapid evaluation, as compressive processes such as epidural or subdural hematoma may need immediate surgical intervention. Once these have been ruled out, however, the underlying traumatic brain injury may itself be sufficient to cause coma.
Traumatic brain injury that causes coma falls into two broad classes: closed head trauma and direct brain injury as a result of penetrating head trauma. Penetrating head trauma
Specific Causes of Structural Coma |
159 |
may directly injure the ascending arousal system, or it may lead to hemorrhage or edema that further impairs brain function. These issues have been discussed in Chapter 3. An additional consideration is that trauma sufficient to cause head injury may also involve the neck, with dissection of a carotid or vertebral artery. These considerations are covered in the sections on vascular occlusions. The discussion that follows will focus primarily on the injuries that occur to the brain as a result of closed head trauma.
Mechanism of Brain Injury During
Closed Head Trauma
During closed head trauma, several physical forces may act upon the brain to cause injury. If the injuring force is applied focally, the skull is briefly distorted and a shock wave is transmitted to the underlying brain. This shock wave can be particularly intense when the skull is struck a glancing blow by a high-speed projectile, such as a bullet. As demonstrated in Patient 3–2, the bullet need not penetrate the skull or even fracture the bone to transmit enough kinetic energy to injure the underlying brain.
A second mechanism of injury occurs when the initial blow causes the head to snap backward or forward, to the point where it is stopped either by the limits of neck movement or by another solid object (a wall or floor, a head restraint in a car, etc.). The initial blow causes the skull to accelerate against the underlying brain, which floats semi-independently in a pool of CSF. The brain then accelerates to the same speed as the skull, but when the skull’s trajectory is suddenly stopped, the brain continues onward to strike the inner table of the skull opposite the original site of the blow (Figure 4–12). This coup-contrecoup injury model was first described by Courville (1950) and then documented in the pioneering studies by Gurdjian,224 who used high-speed motion pictures to capture the brain and skull movements in monkeys in whom the calvaria had been replaced by a plastic dome. If the initial blow is occipital, frontal and temporal lobe damage may be worse than the damage at the site of the blow because of the conformation of the skull, which is smoothly curved at the occipital pole but comes to a narrow angle at
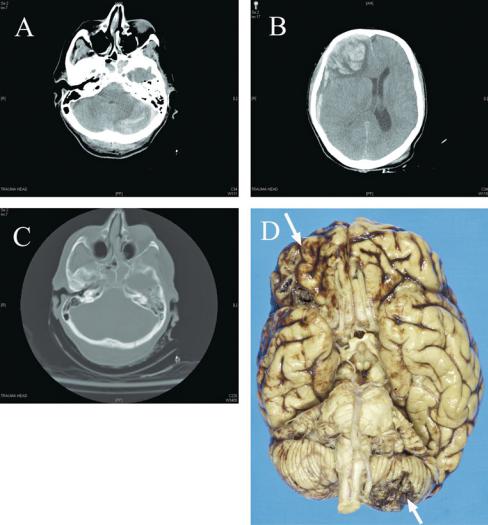
160 Plum and Posner’s Diagnosis of Stupor and Coma
Figure 4–12. A series of computed tomography (CT) scans, and postmortem brain examination, of a 74-year-old woman who fell down a flight of stairs. She was initially alert and confused, but rapidly slipped into coma, which progressed to complete loss of brainstem reflexes by the time she arrived at the hospital. CT scan showed left cerebellar contusion (A) underlying an occipital fracture (C). There was a right frontal intraparenchymal hematoma and subdural hematoma (B). The cerebellar and frontal contusions could be seen from the surface of the brain at autopsy to demonstrate a coup (occipital injury) and contrecoup (frontal contusion from impact against the inside of the skull) injury pattern (arrows in D).
the frontal and temporal poles. As a result of this anatomy, it is not unusual for the greatest damage to the brain to occur at these poles, regardless of where the head is hit. Even in the absence of parenchymal brain damage, movement of the brain may shear off the delicate olfactory nerve fibers exiting the skull through the cribriform plate, causing anosmia.
Brain injury as a result of closed head trauma may be either a contusion (an area of brain
edema visualized on CT or MRI) or focal hemorrhage. Even when no hemorrhage is seen initially on scan, it is not unusual for CSF to show some blood if a lumbar puncture is done. The hemorrhage itself is typically not large enough to cause brain injury or dysfunction. However, the blood may incite seizure activity. Seizures occurring at the time of the head injury do not necessarily herald a subsequent seizure disorder. Nevertheless, seizures themselves and the
following postictal state may complicate the evaluation of the degree of brain injury.
A third mechanism of brain injury is due to shearing force on long axonal tracts. Because the long axis of the brainstem is located at about an 80-degree angle with respect to the long axis of the forebrain, the long tracts connecting the forebrain with the brainstem and spinal cord take an abrupt turn at the mesodiencephalic junction. In addition, because the head is tethered to the neck, which is not displaced by a blow to the head, there is an additional rotational displacement of the head, depending on the angle of the blow. These movements of the forebrain with respect to the brainstem produce a transverse sheering force at the mesodiencephalic juncture, resulting in diffuse axonal injury to the long tracts that run between the forebrain and brainstem.225–228
Mechanism of Loss of
Consciousness in Concussion
The term concussion refers to transient alteration in mental status that may or may not
involve loss of consciousness, resulting from trauma to the brain.229–231 Although the most
dramatic symptom of concussion is transient coma, the hallmarks of the disorder are amnesia and confusion; other symptoms may include headache, visual disturbances, and dizziness.
The mechanism of loss of consciousness with a blow to the head is not completely understood. However, in experiments by Gennarelli and colleagues, using an apparatus to accelerate the heads of monkeys without skull impact, rotational acceleration in the sagittal plane typically produced only brief loss of consciousness, whereas acceleration from the lateral direction caused mainly prolonged and severe coma.227 Brief loss of consciousness, which in humans is usually not associated with any changes on CT or MRI scan, may be due to the shearing forces transiently applied to the ascending arousal system at the mesodiencephalic junction. Physiologically, the concussion causes abrupt neuronal depolarization and promotes release of excitatory neurotransmitters. There is an efflux of potassium from cells with calcium influx into cells and sequestration in mitochondria leading to impaired oxidative metabolism. There are also alterations in cerebral blood flow and glucose metabo-
Specific Causes of Structural Coma |
161 |
lism, all of which impair neuronal and axonal function.231
Longer term loss of consciousness may be due to mechanical injury to the brain, a condition that Adams and colleagues termed diffuse axonal injury.225 Examination of the brains of animals with prolonged unconsciousness in the Gennarelli experiments was associated with diffuse axonal injury (axonal retraction balls and microglial clusters in the white matter, indicating a site of injury) and with hemorrhagic injury to the corpus callosum and to the dorsal surface of the mesopontine junction. These sites underlie the free edge of the falx and the tentorium, respectively. Hence, in these cases the brain displacement is presumably severe enough to hammer the free dural edges against the underlying brain with sufficient force to cause local tissue necrosis and hemorrhage. Similar pathology was seen in 45 human cases
of traumatic closed head injury, all of whom died without awakening after the injury.225,226
Contusion or hemorrhage into the corpus callosum or dorsolateral mesopontine tegmentum may be visible on MRI scan, but diffuse axonal injury generally is not. Magnetic resonance spectroscopy may be useful in evaluating patients with diffuse axonal injury, who typically have a reduction in N-acetylaspartate as well as
elevation of glutamate/glutamine and choline/ creatinine ratios.232–234
Delayed Encephalopathy
After Head Injury
In some cases after an initial period of unconsciousness after a closed head injury, the patient may awaken and the CT scan may be normal, but then the patient may show cognitive deterioration and lapse into coma hours to several days later. This pattern was character-
ized by Reilly and colleagues as patients who ‘‘talk and die.’’10,235 Repeat CT scan typically
shows areas of intraparenchymal edema and perhaps hemorrhage, which may have shown only minimal injury at the time of initial presentation. However, with the evolution of brain edema over the next few hours and days, the mass effect may reach a critical level at which it impairs cerebral perfusion or causes brain herniation.
This condition occurs most commonly in children and young adults in whom the brain
162 Plum and Posner’s Diagnosis of Stupor and Coma
usually fully occupies the intracranial space, so that even minimal swelling may put the brain at risk of injury. Elderly individuals, in whom there has been some cerebral atrophy, may have enough excess intracranial capacity to avoid reaching this crossroad. On the other hand, older individuals may be more likely to deteriorate later due to subdural or epidural hemorrhage or to injuries outside the nervous system.10 Hence, any patient with deterioration of wakefulness in the days following head injury requires repeat and urgent scanning, even if the original scan was normal.
More common is the so-called postconcussion syndrome. This disorder is characterized by headache, dizziness, irritability, and difficulty with memory and attention after mild concussion and particularly after repeated concussions.236 Because it often follows mild head injury, psychologic factors have been imputed by some, but the syndrome clearly appears to result from mild although not anatomically identifiable brain damage.237
INFRATENTORIAL DESTRUCTIVE LESIONS
Infratentorial destructive lesions causing coma include hemorrhage, tumors, infections, and infarcts in the brainstem. Although hemorrhage into tumors, infections, or masses also compress normal tissue, they appear to have their major effect in the brainstem through direct destruction of arousal systems.
If the lesion is large enough, patients with destructive infratentorial lesions often lose consciousness immediately, and the ensuing coma is accompanied by distinctive patterns of respiratory, pupillary, oculovestibular, and motor signs that clearly indicate whether it is the tegmentum of the midbrain, the rostral pons, or the caudal pons that initially is most severely damaged. The brainstem arousal system lies so close to nuclei and pathways influencing the pupils, eye movements, and other major functions that primary brainstem destructive lesions that cause coma characteristically cause focal neurologic signs that can precisely localize the lesion anatomically. This restricted, discrete localization is unlike metabolic lesions causing coma, where the signs commonly indicate incomplete but symmetric dysfunction and few, if any, focal signs of brainstem dysfunction
(see Chapter 2). Primary brainstem injury also is unlike the secondary brainstem dysfunction that follows supratentorial herniation, in which all functions above a given brainstem level tend to be lost as the process descends from rostral to caudal along the neuraxis.
Certain combinations of signs stand out prominently in patients with infratentorial destructive lesions causing coma. At the midbrain level, centrally placed brainstem lesions interrupt the pathway for the pupillary light reflex and often damage the oculomotor nuclei as well. The resulting deep coma commonly is accompanied by pupils that are fixed at midposition or slightly wider, by abnormalities of eye movements due to damage to the third or fourth nerves or their nuclei, and by long-tract motor signs. These last-mentioned signs result from involvement of the cerebral peduncles and commonly are bilateral, although asymmetric.
Destructive lesions of the rostral pons commonly spare the oculomotor nuclei but interrupt the medial longitudinal fasciculus and the adjacent ocular sympathetic pathways. Patients typically have tiny pupils, internuclear ophthalmoplegia (only lateral movements of the eyes on vestibulo-ocular testing), and, in many instances, cranial nerve signs of trigeminal or facial dysfunction, betraying pontine destruction.
Severe midpontine destruction can cause a functional transection with physiologic effects that may be difficult to differentiate from metabolic coma. The pupils of such patients are miotic but may react minimally to light since midbrain parasympathetic oculomotor fibers are spared. Reflex lateral eye movements are absent because the pontine structures for lateral conjugate eye movements are destroyed. However, upward and downward ocular deviation occasionally is retained either spontaneously or in response to vestibulo-ocular testing, and if present, this dissociation between lateral and vertical movement clearly identifies pontine destruction. Ocular bobbing sometimes accompanies such acute destructive lesions and when present usually, but not always, indicates primary posterior fossa disease. The motor signs of severe pontine destruction are not the same in every patient and can include flaccid quadriplegia, less often extensor posturing, or occasionally extensor posturing responses in the arms with flexor responses or flaccidity in the legs. Respiration may show any of the patterns

characteristic of low brainstem dysfunction described in Chapter 1, but cluster breathing, apneusis, gasping, and ataxic breathing are characteristic.
As discussed in Chapter 2, patients with destructive lesions confined to the lower pons or medulla do not show loss of consciousness, although they may be locked in, in which case only the preservation of voluntary vertical eye and eyelid movements may indicate the wakeful state.
BRAINSTEM VASCULAR DESTRUCTIVE DISORDERS
In contrast to the carotid circulation, occlusion of the vertebrobasilar system is frequently associated with coma. Although lesions confined to the lower brainstem do not cause coma, impairment of blood flow in the vertebral or low basilar arteries may reduce blood flow distally in the basilar artery to a level that is below the critical minimum necessary to maintain normal function. The classic presentation of ischemic coma of brainstem origin is produced by occlusion of the basilar artery. The patient falls acutely into a comatose state, and the pupils may initially be large, usually indicating intense adrenal outflow at the time of the initial onset, but eventually become either miotic (pontine level occlusion) or fixed and midposition (midbrain level occlusion). Oculovestibular eye movements may be absent, asymmetric, or skewed (pontine level), or vertical and adduction movements may be absent with preserved abduction (midbrain level). There may be hemiplegia, quadriplegia,ordecerebrateposturing.Respiration may be apneustic or ataxic in pattern if the lesion also involves the pons.
Occlusion of the basilar artery either by thrombosis or embolism is a relatively common cause of coma. The occlusions are usually the result of atherosclerotic or hypertensive disease. Emboli to the basilar artery usually result from valvular heart disease or artery-to-artery embolization.238 Cranial arteritis involving the vertebral arteries in the neck also can lead to secondary basilar artery ischemia with brainstem infarction and coma.239 Vertebrobasilar artery dissection, either from trauma such as whiplash injury or chiropractic manipulation240 or occurring spontaneously, is becoming increasingly recognized as a common cause of
Specific Causes of Structural Coma |
163 |
brainstem infarction, due to the ease of identifying it on MR angiography.241
Most patients in coma from brainstem infarction are over 50 years of age, but this is not an exclusive limit. One of our patients was only 34 years old. The onset can be sudden coma or progressive neurologic symptoms culminating in coma. In some patients, characteristic transient symptoms and signs owing to brief ischemia of the brainstem precede coma by days or weeks.242 These transient attacks typically change from episode to episode but always reflect infratentorial CNS dysfunction and include headaches (mainly occipital), diplopia, vertigo (usually with nausea), dysarthria, dysphagia, bilateral or alternating motor or sensory symptoms, or drop attacks (sudden spontaneous falls while standing or walking, without loss of consciousness and with complete recovery in seconds). The attacks usually last for as short a period as 10 seconds or as long as several minutes. Seldom are they more prolonged, although we have seen recurrent transient attacks of otherwise unexplained akinetic coma lasting 20 to 30 minutes in a patient who later died from pontine infarction caused by basilar occlusion. Except in patients who additionally have recurrent asystole or other severe cardiac arrhythmias, transient ischemic attacks caused by vertebrobasilar artery insufficiency nearly always occur in the erect or sitting position. Some patients with a critical stenosis may have positional symptoms, which are present while sitting but improve when lying down.
Patient 4–5
A 78-year-old architect with hypertension and diabetes was returning on an airplane from Europe to the United States when he complained of dizziness, double vision, and nausea, then collapsed back into his seat unconscious. His seat was laid back and he gradually regained consciousness. A neurologist was present on the airplane and was called to his side. Limited neurologic examination found that he was drowsy, with small but reactive pupils and lateral gaze nystagmus to either side. There was dysmetria with both hands.
On taking a history, he was returning from a vacation in Germany where he had similar symptoms and had been hospitalized for several weeks.

164 Plum and Posner’s Diagnosis of Stupor and Coma
MRI scans, which he was carrying with him back to his doctors at home, showed severe stenosis of the midportion of the basilar artery. He had been kept at bedrest with the head of the bed initially down, but gradually raised to 30 degrees while in the hospital, and then discharged when he could sit without symptoms. His chair back was kept as low as possible for the remainder of the flight, and he was taken from the airplane to a tertiary care hospital where he was treated with anticoagulants and gradual readjustment to an upright posture.
In some cases, segmental thrombi can occlude the vertebral or basilar arteries while producing only limited and temporary symptoms of brainstem dysfunction.242 In one series, only 31 of 85 patients with angiographically proved basilar or bilateral vertebral artery occlusion were stuporous or comatose.242 The degree of impairment of consciousness presumably depends on how much the collateral vascular supply protects the central brainstem
structures contributing to the arousal system. The clinical signs of basilar artery occlusion are listed in Table 4–15. Most unconscious patients have respiratory abnormalities, which may include periodic breathing, or various types of irregular or ataxic respiration. The pupils are almost always abnormal and may be small (pontine), midposition (midbrain), or dilated (third nerve outflow in midbrain). Most patients have divergent or skewed eyes reflecting direct nuclear and internuclear damage (Table 4–15). Patients with basilar occlusion who become comatose have a nearly uniformly fatal outcome in the absence of thrombolytic or endovascular intervention.243
The diagnosis can usually be made on the basis of clinical signs alone, and eye movement signs are particularly helpful in determining the brainstem level of the dysfunction (Table 4–16). However, the nature of the problem must be confirmed by imaging.
Acutely, the CT scan may not reveal a parenchymal lesion. Occasionally, hyperintensity
Table 4–15 Symptoms and Signs of Basilar Artery Occlusion in 85 Patients
|
No. of |
Symptom |
Patients |
|
|
Vertigo, nausea |
39 |
Headache, neckache |
22 |
Dysarthria |
23 |
Ataxia, dysdiadochokinesia |
27 |
Cranial nerve palsy |
|
III |
13 |
IV, VI, VII |
30 |
VIII (acoustic) |
5 |
IX-XII |
24 |
Occipital lobe signs |
11 |
Respiration |
9 |
Central Horner’s syndrome |
4 |
Seizures |
4 |
Sweating |
5 |
Myoclonus |
6 |
|
|
|
No. of |
Consciousness |
Patients |
|
|
Awake |
31 |
Psychosis, disturbed memory |
5 |
Somnolence |
20 |
Stupor |
5 |
Coma |
26 |
|
|
No. of |
|
Long Tract Signs |
Patients |
|
|
|
|
Hemiparesis |
21 |
|
Tetraparesis |
31 |
|
Tetraplegia |
15 |
|
Locked-in syndrome |
9 |
|
Hemihypesthesia |
11 |
|
|
|
|
Supranuclear Oculomotor |
No. of |
|
Disturbances |
Patients |
|
|
|
|
Horizontal gaze paresis |
22 |
|
Gaze-paretic, gaze-induced |
15 |
|
nystagmus |
|
|
Oculocephalic reflex lost |
6 |
|
Vestibular nystagmus |
5 |
|
Vertical gaze palsy |
4 |
|
Downbeat nystagmus |
4 |
|
Internuclear ophthalmoplegia |
4 |
|
Ocular bobbing |
3 |
|
One-and-a-half syndrome |
2 |
|
Other/not classifiable |
16 |
Modified from Ferbert et al.242

Specific Causes of Structural Coma |
165 |
Table 4–16 Eye Movement Disorders in Brainstem Infarcts
Midbrain Syndromes |
Pons Syndromes |
|
|
Upper Midbrain Syndromes |
Paramedian Syndromes |
Conjugate vertical gaze palsy: upgaze palsy, |
Conjugate disorders |
downgaze, palsy, combined upgaze and |
Ipsilateral gaze paralysis |
downgaze palsy |
Complete gaze paralysis |
Dorsal midbrain syndrome |
Loss of ipsilateral horizontal saccades |
Slowness of smooth pursuit movements |
Loss of both horizontal and vertical saccadic |
Torsional nystagmus |
gaze movements |
Pseudoabducens palsy |
Primary-position downbeating nystagmus |
Convergence-retraction nystagmus |
Tonic conjugate eye deviation away from lesion |
Disconjugate vertical gaze palsy: monocular |
from lesion |
elevation palsy, prenuclear syndrome of the |
Disconjugate disorders |
oculomotor nucleus, crossed vertical gaze |
Unilateral internuclear ophthalmoplegia |
paresis, and vertical one-and-a half |
Bilateral internuclear ophthalmoplegia |
syndrome |
Internuclear ophthalmoplegia and |
Skew deviation with alternating appearance |
skew deviation |
Ocular tilt reaction |
One-and-a-half syndrome |
See-saw nystagmus |
Paralytic pontine exotropia |
Middle Midbrain Syndrome |
Ocular bobbing: typical, atypical, and paretic |
Nuclear third nerve palsy |
Lateral Pontine Syndrome |
Fascicular third nerve palsy: isolated or |
Horizontal gaze palsy |
associated with crossed hemiplegia, |
Horizontal and rotatory nystagmus |
ipsilateral or contralateral hemiataxia, |
Skew deviation |
and abnormal movements |
Internuclear ophthalmoplegia |
Lower Midbrain Syndrome |
Ocular bobbing |
Internuclear ophthalmoplegia: isolated or |
One-and-a-half syndrome |
associated with fourth nerve palsy, bilateral |
|
ataxia, and dissociated vertical nystagmus |
|
Superior oblique myokymia |
|
|
|
Modified from Moncayo and Bogousslavsky.244 |
|
within the basilar artery on CT will suggest basilar occlusion.245 The best diagnostic test is an MRI scan with diffusion-weighted imaging (see Figure 4–9B). Early diagnosis may allow effective treatment with thrombolysis,246 angioplasty,247 or embolectomy.248 The differential diagnosis of acute brainstem infarction can usually be made from clinical clues alone. With brainstem infarction, the fact that signs of midbrain or pontine damage accompany the onset of coma immediately places the site of the lesion as infratentorial. The illness is maximal at onset or evolves rapidly and in a series of steps, as would be expected with ischemic vascular disease. Supratentorial ischemic vascular lesions, by contrast, with rare exceptions pointed out on page 152, are not likely to cause coma at onset, and they do not begin with pupillary abnormalities or other signs of direct brainstem injury (unless the mesencephalon is also
involved, e.g., as in the top of the basilar syndrome). Pontine and cerebellar hemorrhages, since they also compress the brainstem, sometimes resemble brainstem infarction in their manifestations. However, most such hemorrhages have a distinctive picture (see above). Furthermore, they nearly always arise in hypertensive patients and often are more likely to cause occipital headache (which is unusual with infarction).
Patient 4–6
A 56-year-old woman was admitted in coma. She had been an accountant and in good health, except for known hypertension treated with hydrochlorothiazide. She suddenly collapsed at her desk and was rushed to the emergency department,