

Книги по МРТ КТ на английском языке / PLUM AND POSNER S DIAGNOSIS OF STUPOR AND COM-1
.pdf196 Plum and Posner’s Diagnosis of Stupor and Coma
and abducted (i.e., ‘‘stopping traffic’’). Incipient asterixis comprises a slight irregular tremor of the fingers, beginning after a latent period of 2 to 30 seconds that is difficult to distinguish from the tremor of metabolic encephalopathy. Leavitt and Tyler45 have described the two separate components of this tremulousness. One is an irregular oscillation of the fingers, usually in the anterior-posterior direction but with a rotary component at the wrist. The second consists of random movements of the fingers at the metacarpal-phalangeal joints. This second pattern becomes more and more marked as the patient holds his or her wrist dorsiflexed until fi- nally the fingers lead the hand into a sudden downward jerk followed by a slower return to the original dorsiflexed position. Both hands are affected, but asynchronously, and as the abnormal movement intensifies, it spreads to the feet, tongue, and face (dorsiflexion of the feet is often an easier posture for obtunded patients to maintain). Indeed, with severe met-
abolic tremors it sometimes becomes difficult to distinguish between intense asterixis and myoclonus, and there is some evidence that the two types of movements represent the same underlying phenomena (sudden and transient loss of muscle tone followed by sudden compensation). Asterixis is generally seen in awake but lethargic patients and generally disappears with the advent of stupor or coma, although occasionally one can evoke the arrhythmic contraction in such subjects by passively dorsiflexing the wrist. Asterixis can also be elicited in stuporous patients by passively flexing and abducting the hips.46 Flapping abductionadduction movements occurring either synchronously or asynchronously suggest metabolic brain disease (Figure 5–1).
Unilateral, or less commonly bilateral, asterixis has been described in patients with focal brain lesions.43 Electromyograms recorded during asterixis show a brief absence of muscular activity during the downward jerk followed by
Figure 5–1. (A) Technique of hip flexion-abduction. (B) Electromyographic (EMG) recording from the hip adductors (upper trace) and accelerometric recording from the patella (lower trace). Brief periods of EMG silence (black dots) are followed by a burst of high-voltage electrical activity and a striking change in acceleration. (From Noda et al.,46 with permission.)
Multifocal, Diffuse, and Metabolic Brain Diseases Causing Delirium, Stupor, or Coma |
197 |
a sudden muscular compensatory contraction, much like the sudden bobbing of the head that normally accompanies drowsiness. The sudden
electrical silence is unexplained and not accompanied by EEG changes.42,45,47
Multifocal myoclonus consists of sudden, nonrhythmic, nonpatterned gross twitching involving parts of muscles or groups of muscles first in one part of the body, then another, and particularly affecting the face and proximal limb musculature. Multifocal myoclonus most commonly accompanies uremic encephalopathy, a large dose of intravenous penicillin, CO2 narcosis, and hyperosmotic-hyperglycemic encephalopathy. Multifocal myoclonus, in a patient who is stuporous or in coma, is indicative of severe metabolic disturbance. However, it may be seen in some waking patients with neurodegenerative disorders (e.g., Lewy body dementia or Alzheimer’s disease) or prion disorders (Creutzfeldt-Jakob disease and related disorders). Its physiology is unknown; the motor twitchings are not always reflected by a specific EEG abnormality and have, in fact, been reported in a patient with electrocerebral silence.48
DIFFERENTIAL DIAGNOSIS
Distinction Between Metabolic
and Psychogenic Unresponsiveness
In awake patients, differences in the mental state, the EEG, the motor signs, and, occasionally, the breathing pattern distinguish metabolic from psychiatric disease. Most conscious patients with metabolic brain disease are confused and many are disoriented, especially for time. Their abstract thinking is defective; they cannot concentrate well and cannot easily retain new information. Early during the illness, the outstretched dorsiflexed hands show irregular tremulousness and, frequently, asterixis. Snout, suck, and grasp reflexes are seen. The EEG is generally slow. Posthyperventilation apnea may be elicited and there may be hypoventilation or hyperventilation, depending on the specific metabolic illness. By contrast, awake patients with psychogenic illness, if they will cooperate, are not disoriented and can retain new information. If they seem disoriented, they are disoriented to self (i.e., they report
that they don’t know who they are) as well as to time and place; disorientation to self almost never occurs in delirious patients. They also lack abnormal reflexes or adventitious movements, although they may have irregular tremor, and they have normal EEG frequencies. Ventilatory patterns, with the exception of psychogenic hyperventilation, are normal.
Unresponsive patients with metabolic disease have even slower activity in their EEGs than responsive patients with metabolic disease, and caloric vestibulo-ocular stimulation elicits either tonic deviation of the eyes or, if the patient is deeply comatose, no response. Psychogenically unresponsive patients have normal EEGs and a normal response to caloric irrigation, with nystagmus having a quick phase away from the side of ice water irrigation; there is little or no tonic deviation of the eyes (see page 65). In some patients with psychogenic coma, the eyes deviate toward the ground when the patient is placed on his or her side.49 Forced downward deviation of the eyes has been described in patients with psychogenic seizures.50
Distinction Between Coma of
Metabolic and Structural Origin
As discussed in Chapter 2, the key to distinguishing coma of metabolic versus structural origin is to identify focal neurologic signs that distinguish structural coma. On the other hand, certain characteristic motor and EEG findings can help confirm the diagnosis of a metabolic encephalopathy when patients are merely obtunded or lethargic. Most patients with metabolic brain disease have diffusely abnormal motor signs including tremor, myoclonus, and, especially, bilateral asterixis. The EEG is diffusely, but not focally, slow. The patient with gross structural disease, on the other hand, generally has abnormal focal motor signs and if asterixis is present, it is unilateral. The EEG may be slow, but in addition with supratentorial lesions, a focal abnormality will usually be present.
Finally, metabolic and structural brain diseases are distinguished from each other by a combination of signs and their evolution. Comatose patients with metabolic brain disease usually suffer from partial dysfunction affecting many levels of the neuraxis simultaneously, yet concurrently retain the integrity of other
198 Plum and Posner’s Diagnosis of Stupor and Coma
functions originating at the same levels. The orderly rostral-caudal deterioration that is characteristic of supratentorial mass lesions does not occur in metabolic brain disease, nor is the anatomic defect regionally restricted as it is with subtentorial damage.
ASPECTS OF CEREBRAL
METABOLISM PERTINENT
TO COMA
Earlier chapters of this book have described the physiologic relationships among the brainstem, the diencephalon, and the cerebral hemispheres that underlie the wakeful state and normally generate the psychologic activities that constitute full consciousness. The brain’s sensorimotor and mental activities are closely coupled to cerebral metabolism so that neurochemical impairment or failure from any cause is likely to produce rapidly evolving neurologic abnormalities.
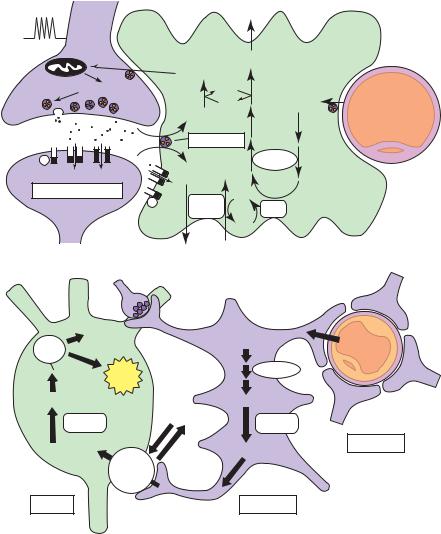
Neurons and glial cells undergo many chemical processes in fulfilling their specialized functions. The nerve cells must continuously maintain their membrane potentials, synthesize and store transmitters, manufacture axoplasm, and replace their always decaying structural components (Figure 5–2). Glia, which constitute 90% of the brain’s cells, have several functions, some of which have been recently recognized.51,52 The oligodendroglial cells have as their major role the generation and maintenance of myelin. Microglia (macrophages) are the brain’s immune cells. Astrocytes regulate much of the ion homeostasis of the brain’s extracellular fluid. In addition, they may aid neuronal function by supplying substrate (lactate)51 (although the degree, if any, to which neurons metabolize lactate in vivo is controversial53). Astrocytes also participate in controlling blood flow52 and in maintaining the blood-brain barrier.54 All of these complex activities require energy, in fact, more of it per kilogram weight of cells than in any other organ in the body. Furthermore, many of the enzymatic reactions of both neurons and glial cells, as well as of the specialized cerebral capillary endothelium, must be catalyzed at some point by the energy-yielding hydrolysis of adenosine triphosphate (ATP) to adenosine diphosphate (ADP) and inorganic phosphate. Without a constant and generous supply of
ATP, cellular synthesis slows or halts, neuronal functions decline or cease, and cell structures quickly crumble.
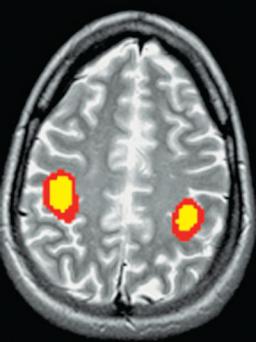
Oxygen, glucose, and cerebral blood flow (CBF) operate interdependently to supply the brain with the substrate and cofactors it requires to carry out the chemical reactions that generate its energy and synthesize its structural components. Awake or asleep, the brain metabolizes at one of the highest rates of any organ in the body. However, although the overall metabolism of the brain is relatively constant, different areas of the brain metabolize at different rates, depending on how active an area is.55 For example, during exercise, the activity of the motor cortex increases dramatically, compensated for by decreased metabolism elsewhere in the brain.56 Changes in regional metabolism are best demonstrated by functional magnetic resonance imaging (MRI) or positron emission tomography (PET) imaging (Figure 5–3). The brain suffers a special vulnerability in that it possesses almost no reserves of its critical nutrients, so that even a brief interruption of blood flow or oxygen supply threatens the tissue’s vitality. These considerations are central to an understanding of many of the metabolic encephalopathies, and the following paragraphs discuss them in some detail.
CEREBRAL BLOOD FLOW
Under normal resting conditions, the total CBF in man is about 55 mL/100 g/minute, an amount that equals 15% to 20% of the resting cardiac output. A number of studies have found that the overall CBF remains relatively constant during the states of wakefulness or slow-wave sleep as well as in the course of various mental and physical activities. PET and functional MRI scanning reveal that this apparent uniformity masks a regionally varying and dynamically fluctuating CBF, which is closely adjusted to meet the metabolic requirements posed by local physiologic changes in the brain. Overall flow in gray matter, for example, is normally three to four times higher than in white matter.55
When neural activity increases within a region, cerebral metabolism increases to meet the increased demand.57 Cerebral metabolic rate for glucose and CBF each increase about 50% in the active area, whereas the metabolic rate for oxygen increases only about 5%.57
A
glutamatergic synapse |
|
astrocyte |
|
capillary |
|
|
|
|
|
Vm
2 LACTATE
GLUTAMINE
glutamate |
ADP |
ATP |
GLUCOSE |
GLUCOSE |
GLUTAMATE
|
EAAT 1 & 2 |
|
G |
|
glycolysis |
Na+ Ca2+ |
3 Na+ |
|
glutamate receptors |
Na+/K+ |
ATP |
G |
||
|
ATPase |
PGK |
|
|
ADP |
|
2 K+ |
|
B
|
glutamate |
|
|
|
TCA |
aspartale |
|
GLUCOSE |
GLUCOSE |
cycle |
GABA |
|
|
|
|
|
|
|
|
|
energy |
|
glycolysis |
|
|
|
|
|
|
PYRUVATE |
|
PYRUVATE |
|
|
|
LDH1 |
LACTATE |
LDH5 |
|
|
|
|
||
LACTATE |
|
LACTATE |
capillary |
|
|
|
|||
|
MCT |
|
|
|
|
1 & 2 |
|
|
|
neuron |
|
|
astrocyte |
|
Figure 5–2. (A) Schematic representation of the mechanism for glutamate-induced glycolysis in astrocytes during physiologic activation. At glutamatergic synapses, presynaptically released glutamate depolarizes postsynaptic neurons by acting at specific receptor subtypes. The action of glutamate is terminated by an efficient glutamate uptake system located primarily in astrocytes. Glutamate is cotransported with Naþ, resulting in an increase in the intra-astrocytic concentration of Naþ, leading to an activation of the astrocyte Naþ/Kþ-ATPase. Activation of the Naþ/Kþ-ATPase stimulates glycolysis (i.e., glucose use and lactate production). Lactate, once released by astrocytes, can be taken up by neurons and serves them as an adequate energy substrate. (For graphic clarity, only lactate uptake into presynaptic terminals is indicated. However, this process could also occur at the postsynaptic neuron.) This model, which summarizes in vitro experimental evidence indicating glutamate-induced glycolysis, is taken to reflect cellular and molecular events occurring during activation of a given cortical area. (B) Schematic representation of the proposed astrocyte-neuron lactate shuttle. Following neuronal activation and synaptic glutamate release, glutamate reuptake into astrocytes triggers increased glucose uptake from capillaries via activation of an isoform of the Naþ/Kþ-ATPase, which is highly sensitive to ouabain, possibly the alpha-2 isoform (Pellerin and Magistretti 1994, 1997). Glucose is then processed glycolytically to lactate by astrocytes that are enriched in the muscle form of lactate dehydrogenase (LDH5). The exchange of lactate between astrocytes and neurons is operated by monocarboxylate transporters (MCTs). Lactate is then converted to pyruvate since neurons contain the heart form of LDH (LDH1). Pyruvate, via the formation of acetyl-CoA by pyruvate dehydrogenase (PDH), enters the tricarboxylic acid (TCA) cycle, thus generating 17 adenosine triphosphate (ATP) molecules per lactate molecule. ADP, adenosine diphosphate. (From Magistretti and Pellerin,58 with permission.)
199
200 Plum and Posner’s Diagnosis of Stupor and Coma
Figure 5–3. A functional magnetic resonance imaging scan of the normal individual flexing and extending his fingers. Blood flow increases to a greater degree than oxygen consumption in the motor areas, leading to an increase in oxyhemoglobin. The paramagnetic oxyhemoglobin causes an increased blood oxygen level-dependent signal in the motor cortex bilaterally. (Image courtesy Dr. Andrei Holodny.)
Thus, the oxygen extraction falls, increasing the concentration of oxyhemoglobin in venous blood. This is the basis for the blood oxygenation level dependent (BOLD) signal obtained using functional MRI. The increase in glucose metabolism over oxygen metabolism results in increased lactate production, possibly the substrate for the increased demand of neurons58 (Figure 5–4). The stimulus for the increase in regional CBF is complex.59 A number of vasoactive substances are released by neurons and glia during increased neural activity. Important among these are adenosine, nitric oxide, dopamine, acetylcholine, vasoactive intestinal polypeptide, and arachidonic acid metabolites.59 Several pathologic states of brain are marked by a disproportionately high rate of local blood flow in relation to metabolism. Examples of such reactive hyperemia or ‘‘uncoupling’’ of flow and metabolism occur in areas of traumatic or postischemic tissue injury, as well as in
regions of inflammation or in the regions surrounding certain brain tumors. So far, the nature of the local stimulus to such pathologic vasodilation also has eluded investigators. The effects of the process, however, can act to increase the bulk of the involved tissue and thereby accentuate the pathologic effects of compartmental swelling in the brain, as discussed in Chapter 2.
Reduced CBF has several causes. As described in Chapter 3, the cerebral vasculature’s capacity for autoregulation protects the CBF against all but the most profound drops in systemic blood pressure. The process of autoregulation also means that conditions causing a lowered cerebral metabolism are usually accompanied by a secondary fall in CBF, although in many such cases the initial decline in CBF is less than the metabolic reduction.60 This delayed response may reflect the relatively slow adaptation of the tonic contractile state of vascular smooth muscle rather than a true uncoupling of flow and metabolism. Intrinsic arterial spasm in cerebral vessels, which reduces tissue flow below metabolic needs, is an uncommon phenomenon limited largely to arteries at the base of the brain (e.g., with local surgical trauma as well as with subarachnoid bleeding and sometimes with meningitis [see Chapter 4]). Multifocal cerebral arteriolar spasm had been invoked to explain the regional cerebral vascular injury of malignant hypertension; recent work, however, offers a different interpretation of the pathogenesis of that disorder (see page 168).
Primary reductions in CBF can be regional or general (global). Regional impairments of CBF results from intrinsic diseases of the cervical and cerebral arteries (atherosclerosis, thrombosis, and, rarely, inflammation), from arterial embolism, and from the extrinsic pressure on individual cerebral arteries produced by compartmental herniation. General or global reductions in CBF result from systemic hypotension, complete or functional cardiac arrest (e.g., ventricular arrhythmias in which output falls below requirements of brain perfusion), and increased ICP. As noted earlier in this volume, however, unless some primary abnormality of brain tissue acts to increase regional vascular resistance, an increase in the ICP must approach the systemic systolic pressure before the CBF declines sufficiently to cause recognizable changes in neurologic functions.

Multifocal, Diffuse, and Metabolic Brain Diseases Causing Delirium, Stupor, or Coma |
201 |
|||
|
NO |
|
Astrocytes |
|
|
|
|
|
|
|
VIP |
|
|
|
|
DA |
|
|
|
|
SP |
|
|
|
|
5HT |
|
|
|
Interneuron |
GABA |
|
|
|
NA |
|
|
|
|
central pathways |
|
|
|
|
ACh |
|
P2Y |
|
|
|
|
|
|
|
|
K+ siphoning |
|
tCa2+ |
|
|
Ado ATP |
|
ATP |
|
|
P450 |
|
|
|
|
EETs |
|
Ca2+ |
|
|
Cox2 |
waves |
|
|
|
|
|
||
|
PGs |
tCa2+ 1P3 |
|
|
|
tCa2+ |
|
|
|
|
|
|
Gap |
|
|
|
|
junction |
|
mGluR
H+ K+
Ado NO
PGs
Glu
ATP Ado
NOS tCa2+
Cox2
Figure 5–4. Vasoactive mediators released from neurons and glia by neural activity. Ions (Hþ and Kþ) contribute to the extracellular currents that are associated with synaptic transmission. Adenosine (Ado) is produced through adenosine triphosphate (ATP) catabolism. Glutamate (Glu)-induced increases in the intracellular concentration of Ca2þ in neurons and glia activate the synthesis of nitric oxide (NO), of the cyclooxygenase-2 (Cox2) products prostaglandins (PGs), and of the cytochrome P450 epoxygenase products epoxyeicosatrienoic acids (EETs). In astrocytes, the [Ca2þ] increase is produced by activation of metabotropic glutamate receptors (mGluRs) and by propagation of Ca2þ waves from neighboring astrocytes through activation of purinergic receptors (P2Y) or entry of 1P3 (inositol (1,4,5)-triphosphate) through gap junctions. Astrocytic lipoxygenase products could also produce vasodilation by inducing NO release from endothelial cells. Spatial buffering currents in astrocytes release Kþ from perivascular end-feet, where Kþ conductance is greatest (Kþ siphoning). Interneurons and projecting neurons with perivascular contacts release vasoactive neurotransmitters and neuropeptides, including NO, vasoactive intestinal polypeptide (VIP), dopamine (DA), substance P (SP), serotonin (5HT), gamma-aminobutyric acid (GABA), noradrenaline (NA), and acetylcholine (ACh). (From Iadecola,59 with permission.)
Cessation of blood flow to the brain (ischemia), as discussed in subsequent paragraphs, appears to cause a greater risk of irreversible tissue damage than does even a profound reduction in the arterial oxygen tension (anoxemia). The precise lower level of arterial perfusion required to maintain the vitality of the tissue in man is not known. Extrapolations based on animal experiments suggest that the CBF of
20 mL/100 g of brain per minute causes loss of consciousness but not permanent damage. If the flow falls to 10 mL/100 g/minute, membrane integrity is lost and calcium influx into the cells leads to irreversible damage. Time is also an important factor. Flows of 18 mL can be tolerated for several hours without leading to infarction, whereas flows of 5 mL lasting for more than 30 minutes will cause infarction.61
202 Plum and Posner’s Diagnosis of Stupor and Coma
Several factors may explain why ischemia so severely threatens tissue structure. A change in pH or lactic acid concentration is one factor. Anaerobic metabolism produces large amounts of lactic acid and lowers the pH. The increased concentration of hydrogen ions leads to cell death62 by increasing brain edema, interfering with mitochondrial ATP generation, increasing calcium levels, and the formation of free radicals, all of which can cause cellular death.63 Hypoglycemia (see below), by increasing lactate production, contributes to the brain damage.
Several other factors play a role in helping regulate CBF, the most important of which is PCO2 or, more accurately, cerebral pH. Cerebral acidosis is a potent vasodilator, as is potassium, which leaks into the brain extracellular space during hypoxia. Other factors that serve to increase CBF include nitric oxide (which in older literature was referred to as endothelial-derived relaxing factor), adenosine (probably working through nitric oxide), and prostaglandins (for a review see 59,64).
GLUCOSE METABOLISM
Glucose is the overwhelmingly predominant blood-borne substrate for brain metabolism. One might question why this is so since it is known that slices of cerebral cortex in vitro can utilize a variety of substrates, including fatty acids and other compounds, to synthesize acetoacetate for entry into the citric acid cycle. The answer appears to lie in the specialized properties of the blood-brain barrier, which, by rigorously limiting or facilitating the entry or egress of substances to and from the brain, guards the narrow homeostasis of that organ. Glucose is transported across the blood-brain barrier by a carrier-mediated glucose transporter (Glut-1). The uptake of glucose into neurons is also facilitated by a glucose transporter (Glut-3), and glucose uptake into astrocytes by Glut-1. Under normal circumstances, brain glucose concentration is approximately 30% of that of plasma. Insulin is not required for the entry of glucose into brain or for its metabolism by brain cells. Nevertheless, the brain is rich in insulin receptors with substantial regional variation, the richest area being the olfactory bulb.65 Insulin itself reaches the brain using a transporter that is partially saturated at euglycemic levels. The exact function
of insulin and its receptor in the brain is not known.
In net metabolic terms, each 100 g of brain in a normal human being utilizes about 0.31 mol (5.5 mg) of glucose per minute so that in the basal, prolonged fasting state, the brain’s consumption of glucose almost equals the total amount that the liver produces. This net figure, however, hides the fact that glucose consumption in local regions of the brain varies widely according to local functional changes. Because of its rapid transfer into brain, glucose represents essentially the organ’s only substrate under normal physiologic conditions. However, neurons probably utilize lactate produced from glucose by astrocytes when stimulated with glutamate.66
Ketone bodies can diffuse into brain and also are transported across the blood-brain barrier. These substances provide increased fuel to the brain when beta-hydroxybutyrate, acetoacetate, and other ketones increase in the blood during states such as starvation, the ingestion of high-fat diets, or ketoacidosis. During starvation, in fact, liver gluconeogenesis may fall below the level required to meet cerebral substrate needs; at such times ketone utilization can contribute as much as 30% of the brain’s fuel for oxidative metabolism. For unknown reasons, however, the brain does not appear able to subsist entirely on ketone bodies, and as mentioned below, some investigators believe that ketones contribute to the neurologic toxicity of diabetic ketoacidosis.
Under normal circumstances, all but about 15% of glucose uptake in the brain is accounted for by combustion with O2 to produce H2O and energy, the remainder going to lactate production. The brain contains about 1 mmol/kg of free glucose in reserve and a considerable amount of glycogen, perhaps as high as 10 mg/L, which is present in astrocytes.67,68 With the addition of either increased metabolic demand or decreased metabolic supply, glycogen in astrocytes can break down to lactate to support neuronal function. Despite this, deprivation of glucose and oxygen to the brain rapidly results in loss of consciousness, normal cerebral function being maintained for only a matter of seconds.
The energy balance of the brain is influenced both by its supply of energy precursors (i.e., its input) and by the work the organ does (i.e., its output). Just as intrinsic mechanisms
Multifocal, Diffuse, and Metabolic Brain Diseases Causing Delirium, Stupor, or Coma |
203 |
appropriately increase or decrease the rate of metabolism in different regions of the brain during periods of locally increased or decreased functional activity, intrinsic mechanisms appear able to ‘‘turn down’’ general cerebral metabolic activity and produce stupor or coma when circumstances threaten to deplete blood-borne substrate.
Several metabolic disorders are known to cause a decrease in the brain’s rate of metabolism and physiologic function without initially resulting in any encroachment on the energy reserves of the tissue. The reversible hypometabolism of anesthesia is discussed in a following section. Mechanistically less well understood than anesthesia is a reversible hypometabolism that accompanies the early stages of hypoglycemia, severe hypoxemia, reduced states of CBF, and hyperammonemia. The response appears to be important in protecting the brain against irreversible damage, however, and is well illustrated by describing the neurochemical changes that accompany hypoglycemia. Both hyperglycemia and hypoglycemia can damage the brain.
mend careful control of blood glucose in critically ill patients and those with brain injury of various types.73
The mechanism by which hyperglycemia worsens the prognosis in such patients is not clear. Some believe that the increased production of lactate and lowering of the pH leads to the cellular damage. However, lactate is probably a good substrate for neurons, and the increased blood glucose should be protective. In fact, in experimental animals, a glucose load given 2 to 3 hours before an ischemic insult is protective, but the same glucose load administered 15 to 60 minutes before ischemia aggravates the ischemic outcome,74 although these findings have been challenged.75 Another possible mechanism by which hyperglycemia may damage the brain is that a glucose load leads to release of glucocorticoids that in turn can cause cellular damage.70 Whatever the mechanism, careful control of blood glucose allowing neither hypernor hypoglycemia appears essential for the best care of critically ill and brain-injured patients.
Hyperglycemia
Brain damage from chronic hyperglycemia (i.e., either type 1 or type 2 diabetes) is well established.69 Sustained hyperglycemia causes hyperosmolality, which in turn induces compensatory vasopressin secretion. Although adaptive in the short term, in the long term sustained hyperglycemia damages vasopressin-secreting neurons in the hypothalamus and supraoptic nucleus. In addition, some evidence suggests that sustained hyperglycemia damages hippocampal neurons as well,70 leading to cognitive defects in both humans71 and experimental animals. These effects appear to be independent of diabetes-induced damage to brain vasculature leading to stroke, a common complication of chronic poorly controlled diabetes.
The effect of hyperglycemia on patients with damaged brains is less clear cut. Clinical evidence demonstrates that patients who are hyperglycemic after brain injury, either due to global or focal ischemia72 or to brain trauma, do less well than patients who are euglycemic. The same may well be true for critically ill patients, even those without direct brain damage. These findings have led investigators to recom-
Hypoglycemia
Hypoglycemia deprives the brain of its major substrate and can be expected to interfere with cerebral metabolism by reducing the brain’s energy supply in a manner similar to that caused by hypoxia. With very severe or prolonged hypoglycemia this turns out to be true, but with less severe or transient reductions of glucose availability, one finds that brain function and metabolism decline before one can detect a decline in ATP levels in the tissue.
Soon after insulin came into clinical use, it was realized that hypoglycemic coma could last for up to an hour or so without necessarily leaving any residual neurologic effects or structural brain damage. (This capacity to induce transient but fully reversible coma was important in developing the ineffective76 use of insulin coma in attempts to treat psychiatric disorders.) Since equally long periods of hypoxemic coma always leave neurologic damage in their wake, the difference between the effects of a deficiency of oxygen and a deficiency of substrate has engendered considerable interest. Accordingly, the mechanism of hypoglycemic coma has received repeated attention by biochemists with results important to the
204 Plum and Posner’s Diagnosis of Stupor and Coma
understanding of many aspects of human cerebral metabolism.
Hypoglycemia affects CBF, glucose consumption, and oxygen consumption in different ways. Clinical studies of CBF and metabolism during hypoglycemia in humans indicate that at all levels of blood sugar thus far studied, CBF remains the same or may occasionally rise,77 perhaps from nitric acid release,78 or fall slightly.79 Overall changes in CBF do not reflect regional changes. At modest levels of hypoglycemia (3.0 mmol/L), CBF increases in several areas including the medial prefrontal cortex, whereas it falls in others, such as the hippocampus.79 In an experimental study, a sharp rise in CBF (57%) occurred when blood glucose concentrations fell below 2 mmol/L, at which point brain glucose concentrations approached zero.80 With a relatively mild reduction of blood glucose in humans down to levels of 1.7 to 2.6 mmol/dL (31 to 46 mg/dL), consciousness is preserved, and cerebral glucose consumption (CMRglu) declines moderately but cerebral oxygen consumption remains normal. Despite the preservation of consciousness, at levels of approximately 2.5 mmol/L the latency of the P300 readiness potential increases as does reaction time, suggesting an altered ability to make decisions.81 In patients with hypoglycemic coma, cerebral metabolic rate for oxygen (CMRO2) declines only to an insignificant degree, but CMRglu falls disproportionately by more than half.77 These changes imply that during hypoglycemia the brain is utilizing substrates other than glucose for oxidative metabolism, such as endogenous glycogen82 and lactate.83 Furthermore, despite a normal oxygen consumption, the qualitative change in substrate results in profound functional changes in the neural systems that normally subserve consciousness.
Studies in animals extend the above studies in man and indicate that even with degrees of hypoglycemia sufficient to produce convulsions or deep coma, whole brain energy reserves are at least briefly maintained. Levels of phosphocreatine and ATP remain normal in the brains of mice or rats so long as EEG activity remained. Energy reserves fail only after prolonged convulsive activity or after the EEG becomes isoelectric ( 1 mmol/L84).
Cerebral metabolic studies imply that hypoglycemic confusion, stupor, and even coma in its early stages cannot be attributed simply
to a failure of overall cerebral energy supply. The mechanism by which hypoglycemia causes irreversible neurologic dysfunction is not known, but experimental evidence suggests that impaired acetylcholine metabolism85 or a rise in aspartic acid levels leading to excessive excitation of neurons86 may be involved. Profound hypoglycemia causes pathologic changes in the brain, probably due in part to the massive release of aspartate into the brain extracellular space, flooding excitatory amino acid receptors and causing an influx of calcium, leading to neuronal necrosis.84 Evidence also implicates apoptosis, probably resulting from release of cytochrome C, causing an increase in caspase-like activity.87
Other mechanisms may add to the neurologic dysfunction caused by hypoglycemia. Neurogenic pulmonary edema resulting from a massive sympathetic discharge adds hypoxia to the hypoglycemic insult.88 Transient cerebral edema, either vasogenic89 from increased blood-brain barrier permeability or cytotoxic,90 may also complicate the development of hypoglycemia. A single case report describes the development of central pontine myelolysis (see page 171) associated with hypoglycemic coma, but without electrolyte disturbance, in a patient with anorexia nervosa91; the cause is unknown.
The above discussion on hypoglycemia indicates that the presence or absence of energy failure in the tissue may be the major factor that determines whether cells die or recover. The following section extends the point and compares some of the cerebral metabolic effects of reversible anesthesia with those of anoxic-ischemic and other metabolic conditions producing stupor or coma.
Many directly applied physical and chemical agents can injure the brain. For example, trauma can shear axons and displace tissue sufficiently to cause neuronal death. However, in addition to direct injury, many lethal injuries of the brain exert their effects by producing tissue anoxia. As discussed above, the body normally maintains its nervous tissue in a constant ‘‘high-energy’’ state in which the oxidative metabolism of glucose generates a constant supply of ATP and phosphocreatine to maintain membrane potentials, transmit neuronal impulses, and synthesize protoplasm. When the mechanisms that sustain these energy reserves go awry, ATP and phosphocreatine levels decline, membranes lose their pumping mecha-
Multifocal, Diffuse, and Metabolic Brain Diseases Causing Delirium, Stupor, or Coma |
205 |
nisms, the cells swell, and, at some point, the neuron loses its capacity to recover. Histologic evidence, discussed below, indicates that the mitochondria bear the initial brunt of irreversible damage, while histochemical evidence suggests that oxidative enzymes themselves are destroyed.92 As the precise lethal point of no return is unknown in cellular-molecular terms, one generally must turn to physiologic models when trying to find out just when and why the nervous system dies. Evidence from such models indicates that the brain can harmlessly suspend its activities almost indefinitely when metabolically depressed or cooled, but quickly succumbs when it loses its functional activities in the absence of oxygen or substrate.
ANESTHESIA
General anesthesia and slow-wave sleep are states comparable to pathologic coma, but which maintain normal levels of energy metabolites and are easily reversible. Both may affect the same structures93 in the brain, but the mechanisms of neither are fully understood. In both sleep and anesthesia, there is inhibition of the neuronal pathways making up the ascending arousal system. During sleep, gammaaminobutyric acid (GABA)-ergic neurons in the ventrolateral preoptic nucleus inhibit the components of the ascending arousal system via GABAA receptors (see Chapter 1). Most general anesthetics are potential GABAA receptor agonists that inhibit the activity of the arousal system by activating the same GABAA receptors used by the ventrolateral preoptic nucleus during sleep. The result is slowing of thalamocortical activity in both sleep and general anesthesia.94,95
General anesthetic agents produce immobility and block pain, largely the effect of the anesthetics on the descending brainstem modulatory systems and directly on the spinal cord,
and cause amnesia and loss of consciousness when given in high doses.95,96 These actions for
most anesthetic agents (benzodiazepines, barbiturates, propofol, ethanol, gas anesthetics) are due to activation of different classes of GABAA receptors that contain alpha-1 subunits (most important in sedative and amnestic effects) and alpha-2, -3, and -5 subunits (most important for anxiolytic and muscle relaxant effects).97 Other anesthetic agents, such as nitrous oxide
and ketamine, act mainly as N-methyl-d- aspartate (NMDA) antagonists. These agents distort, rather than depress, thalamocortical activity, and hence are sometimes called dissociative agents rather than anesthetics. Thus, whereas GABAA agonist general anesthetics decrease cerebral metabolism, ketamine increases CBF and maintains oxygen and glucose metabolism at a waking level.98 The depth of anesthesia and the degree of diminution of cerebral metabolism with GABAA agonist anesthetics can be roughly measured by the EEG. As anesthesia deepens, electroencephalographic activity is suppressed; the degree of suppression from none to a completely isoelectric EEG correlates roughly with the cerebral metabolic rate.99 Some general anesthetics actually increase CBF, but they still diminish cerebral metabolism. Thus, clinically, anesthesia depresses the function of the brain but keeps that organ in a high-energy state poised for the resumption of normal function. Well-ventilated animals subject to various concentrations of general anesthetics maintain normal concentrations of ATP and phosphocreatine and normal lactate pyruvate ratios, indicating that no tissue hypoxia has occurred.100 The brain can be depressed to essentially functionless levels by anesthetic depressant drugs, yet lose none of its capacity for total recovery when the anesthetic disappears. Several investigators have demonstrated that experimental animals and humans can be resuscitated to full functional activity after periods of deep anesthesia producing hours to days of isoelectric EEG flattening.101 This tolerability is used clinically in cases of status epilepticus to prevent continuous seizure activity from damaging the brain. A corollary is that in cases of coma due to sedative overdose, the depth and duration of coma are not indicative of the potential for recovery of function.
In animal experiments, general anesthesia, either before or within a few hours of an ischemic insult to the brain, protects against brain damage when measured a few days after the insult. However, at 3 weeks there is no difference in the degree of neuronal damage
between the anesthetized animals and those treated without anesthesia,102,103 indicating no
protection against the delayed effects of anoxia (see page 219).
Clinical experience with barbiturate anesthesia and drug poisoning indicates that given good medical care, most patients usually survive