

Книги по МРТ КТ на английском языке / PLUM AND POSNER S DIAGNOSIS OF STUPOR AND COM-1
.pdfBox 1–3 Wake-Sleep States
In the early days of EEG recording, it was widely assumed that sleep, like coma, represented a period of brain inactivity. Hence, it was not surprising when the EEG appearance of sleep was found to resemble the high-voltage, slow waves that appear during coma. However, in 1953, Aserinsky and Kleitman37 reported the curious observation that, when they recorded the EEG as well as the electromyogram (EMG) and the electro-oculogram (EOG) overnight, their subjects would periodically enter a state of sleep in which their eyes would move and their EEG would appear to be similar to waking states, yet their eyes were closed and they were deeply unresponsive to external stimuli.37,38
This condition of REM sleep has also been called desynchronized sleep (from the appearance of the EEG) as well as paradoxical sleep. More detailed study of the course of a night of sleep revealed that the REM and NREM periods tend to alternate in a rhythmic pattern through the night.39–41
During active wakefulness, the EEG gives the appearance of small, desynchronized waves and the EMG is active, indicating muscle activity associated with waking behavior. In quiet wakefulness, the EEG often begins to synchronize, with 8- to 12-Hz alpha waves predominating, particularly posteriorly over the hemisphere. Muscle tone may diminish as well. As sleep begins, the EEG rhythm drops to the 4- to 7-Hz theta range, muscle tone is further diminished, and slowly roving eye movements emerge (stage I NREM). The appearance of sleep spindles (waxing and waning runs of alpha frequency waves) and large waves in the 1- to 3-Hz delta range, called K complexes, denotes the onset of stage II NREM. The subject may then pass into the deeper stages of NREM, sometimes called slow-wave sleep, in which delta waves become a progressively more prominent (stage III) and then dominant (stage IV) feature. During these periods, eye movements are few and muscle tone drops to very low levels. This usually takes about 45 to 60 minutes, and then the subject often will gradually emerge from the first bout of slow-wave sleep to stage I again.
At this point, the first bout of REM sleep of the night often occurs. The subject abruptly transitions into a desynchronized, low-voltage EEG, with rapid and
Awake |
Sleep stage 1 |
EOG
EMG
EEG
2
3 |
4 |
REM |
50 V
1 s
Figure B1–3A. The main features of a polysomnogram showing the eye movements (electro-oculogram [EOG]), muscle tone (electromyogram [EMG]), and electroencephalogram (EEG) across the different stages of sleep and wakefulness. During wakefulness, the EEG is desynchronized, the EMG is active, and there are spontaneous eye movements. During non-rapid eye movement (NREM) sleep, the EEG becomes progressively slower, the EMG less active, and eye movements slow down or become slowly roving. During REM sleep, there is a rapid transition to a desynchronized EEG, and irregular, rapid eye movements, but the EMG becomes minimal, consistent with atonia. (From Rechtschaffen, A, and Siegel, J. Sleep and dreaming. Chapter 47 in: Kandel, ER, Schwartz, JH, Jessel, TM. Principles of Neural Science. 4th ed. McGraw-Hill, New York, 2000, pp. 936–947. By permission of McGraw-Hill.)
(continued)
16
vigorous eye movements and virtually complete loss of muscle tone, except in the muscles of respiration. The first bout of REM sleep during the night typically lasts only 5 to 10 minutes, and then the subject will transition into stage I NREM, and again begin to descend gradually into deeper stages of NREM sleep.
As the night progresses, the subject typically will spend progressively less time in the deeper stages of NREM sleep, and more time in REM sleep, so that most of the REM sleep for the night comes in the last few bouts. Spontaneous awakenings during the night typically occur from the lighter stages of NREM sleep. Active dreams
Childhood
Awake
REM 1 2 3 4
1 |
2 |
3 |
4 |
5 |
6 |
7 |
Early adulthood
Sleep stages
Awake
REM 



 1 2 3 4
1 2 3 4
1 |
2 |
3 |
4 |
5 |
6 |
7 |
Old age
Awake
REM 1 2 3 4
1 |
2 |
3 |
4 |
5 |
6 |
7 |
|
|
Hours of night |
|
|
|
|
Figure B1–3B. The stages of sleep through the night in a child, young adult, and older person. There is usually regular progression from wakefulness through the stages of non-rapid eye movement (NREM) sleep into its deepest stages, then progression back to light NREM sleep before the first REM episode of the night. With successive cycles through the night, the amount of deeper NREM sleep becomes less, and the amount of REM becomes greater. With aging, the amount of deep NREM sleep diminishes, and sleep fragmentation with more frequent awakenings is seen. (From Rechtschaffen, A, and Siegel, J. Sleep and dreaming. Chapter 47 in: Kandel, ER, Schwartz, JH, Jessel, TM. Principles of Neural Science. 4th ed. McGraw-Hill, New York, 2000, pp. 936–947. By permission of McGraw-Hill.)
(continued)
17

18 Plum and Posner’s Diagnosis of Stupor and Coma
Box 1–3 Wake-Sleep States (cont.)
occur predominantly during REM sleep, although many subjects report passive dreams and ideation during NREM sleep as well.
This pattern, which is typical of young adults, changes dramatically across a lifetime. Infants spend much more time asleep, and much more time in the deeper stages of NREM sleep, than adults. The amount of stages III and IV NREM sleep diminishes as children enter puberty, and it may not occur at all in some older adults. Thus, phenomena such as night terrors, bed wetting, and sleep walking tend to occur mainly during slow-wave sleep in children but disappear as the children become older and spend less time in those sleep stages. Most sedative drugs are GABAA receptor agonists that acutely increase the amount of time spent in the lighter stages of NREM sleep, but there may be little time spent in stages III or IV of NREM or in REM sleep. These drugs are thought to act directly on the arousal system, inhibiting the firing of its neurons. Newer drugs such as gaboxadol, which acts on a specific class of GABAA receptors containing delta subunits, may allow activation of the endogenous sleep system of the brain, and produce a pattern of sleep including more deep slow waves and more REM sleep.
by Lu and Saper (unpublished) have focused on neurons in the mesopontine tegmentum that provide inputs to the basal forebrain, which is critical for maintaining a wakeful state. Populations of neurons in the pre-locus coeruleus area and medial parabrachial nucleus have intense inputs to the basal forebrain. Cell-specific lesions of these neurons produce profound coma, suggesting that they may be a major source of the ascending arousal influence.
In addition, along the course of the ascending cholinergic and monoaminergic axons through the rostral midbrain reticular formation, there are many additional neurons that project to the thalamic relay, midline, and intralaminar nuclei.34 Most of these neurons appear to be glutamatergic, and they may amplify the arousal signal that arises in the mesopontine tegmentum. On the other hand, they do not appear to be capable of maintaining a waking state in the case of acute loss of the influence from the mesopontine neurons.
Along the course of the ascending arousal systems, as they pass through the hypothalamus, are several hypothalamic cell groups that augment the ascending projection to the basal forebrain and cerebral cortex. These include histaminergic neurons in the tuberomammillary nucleus as well as several populations of neurons in the lateral hypothalamic area, all of which project diffusely to the cerebral cortex and
innervate the intralaminar and midline thalamus.54 There is considerable evidence that the histaminergic input in particular is important for maintaining a wakeful state. Histamine H1 blockers impair wakefulness in both animals and humans,55 and transgenic mice lacking H1 receptors have impairment of arousal responses induced by intraventricular injection of the peptide orexin.56 Transgenic mice lacking histidine decarboxylase show a deficit in wakefulness induced by a novel environment, and mice injected with an inhibitor of this key enzyme for histamine synthesis similarly show less wakefulness.57
Some of the lateral hypothalamic neurons contain orexin,58 a peptide that is associated with arousal, and others contain melaninconcentrating hormone59,60 or GABA.61 Many neurons in the lateral hypothalamic area, including those that contain orexin, fire fastest
during wakefulness and slow down during both slow-wave and REM sleep.62,63 Alterna-
tively, the firing of some lateral hypothalamic neurons, which are likely to contain melanin-
concentrating hormone, increases during REM
sleep.38,64,65
In addition, the ascending monoaminergic and hypothalamic projections pass through the basal forebrain, and along their pathway to the cerebral cortex, they encounter and are augmented further by additional populations of
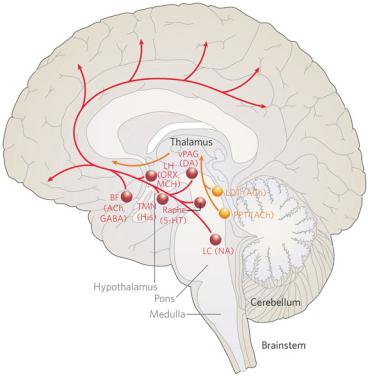
Pathophysiology of Signs and Symptoms of Coma |
19 |
Figure 1–2. A summary diagram of the ascending arousal system. The cholinergic system, shown in yellow, provides the main input to the relay and reticular nuclei of the thalamus from the upper brainstem. This inhibits the reticular nucleus and activates the thalamic relay nuclei, putting them into transmission mode for relaying sensory information to the cerebral cortex. The cortex is activated simultaneously by a series of direct inputs, shown in red. These include monoaminergic inputs from the upper brainstem and posterior hypothalamus, such as noradrenaline (NA) from the locus coeruleus (LC), serotonin (5-HT) from the dorsal and median raphe nuclei, dopamine (DA) from the ventral periaqueductal gray matter (vPAG), and histamine (His) from the tuberomammillary nucleus (TMN); peptidergic inputs from the hypothalamus such as orexin (ORX) and melanin-concentrating hormone (MCH) both from the lateral hypothalamus (LH); and both cholinergic (ACh) and gamma-aminobutyric acid (GABA)-ergic inputs from the basal forebrain (BF). Activation of the brainstem yellow pathway in the absence of the red pathways occurs during rapid eye movement (REM) sleep, resulting in the cortex entering a dreaming state. LDT, laterodorsal tegmental nuclei; PPT, pedunculopontine. (From Saper, CB, Scammell, TE, Lu J. Hypothalamic regulation of sleep and circadian rhythms. Nature 437:1257–1263, 2005. By permission of Nature Publishing Group.)
cholinergic and noncholinergic neurons in the magnocellular basal forebrain nuclei.76 These large cholinergic neurons receive afferents from virtually all of the hypothalamic and monoaminergic brainstem ascending systems and accompany them to diffusely innervate the cerebral cortex.77,78 However, the pattern of termination of the cholinergic neurons is more specific than the monoamine inputs to the cortex. Whereas axons from individual monoaminergic neurons typically ramify widely in the cerebral cortex, axons from basal forebrain cholinergic neurons each innervate a patch of cortex of only a few millimeters in diameter.42,54 Recordings from basal forebrain neurons in rats across the wakesleep cycle indicate that they have a wide range
of activity patterns. Many are most active during wakefulness or during slow-wave sleep, and they fire in bursts that correlate with EEG wave patterns.79 Interestingly, in behaving monkeys, basal forebrain neuron firing correlates best with the reward phase of complex behaviors, suggesting that these neurons may be involved in some highly specific aspect of arousal, such as
focusing attention on rewarding tasks, rather than in the general level of cortical activity.80,81
Thus, the ascending arousal system consists of multiple ascending pathways originating in the mesopontine tegmentum, but augmented by additional inputs at virtually every level through which it passes on its way to the basal forebrain, thalamus, and cerebral cortex. These

Box 1–4 Orexin and Narcolepsy
From its first description by Gelineau in 1880,66 narcolepsy had puzzled clinicians and scientists alike. Although Gelineau included within his definition a wide range of disorders with excessive daytime sleepiness, Gowers has been credited with limiting the term to cases with brief periods of sleep that interrupt a normal waking state. Kinnier Wilson firmly identified it with attacks of cataplexy, during which ‘‘the patient’s knees give way and he may sink to the ground, without any loss of consciousness.’’24 Wilson pointed out that narcolepsy had been considered a very rare condition of which he had seen only a few cases during the first 20 years of his practice, but that in the mid-1920s there was a sudden increase in the number of cases, so that he had seen six within a year in 1927; Spiller reported seeing three within a year in 1926. Wilson opined that the epidemic of new cases of narcolepsy in those years was due to the worldwide epidemic of encephalitis from about 1918 to 1925. However, the prevalence of narcolepsy has remained relatively high, with a current rate of one per 2,000 population, and it has its peak incidence during the second and third decades of life.38
Over the years, additional features of narcolepsy were described. About half of patients reported sleep paralysis, a curious state of inability to move during the transition from sleep to wakefulness or from wakefulness to sleep.38 However, up to 20% of normal individuals may also experience this condition occasionally. More characteristic of narcolepsy, but occurring in only about 20% of cases, are episodes of hypnagogic hallucinations, during which the patient experiences a vivid, cartoon-like hallucination, with movement and action, against a background of wakefulness. The patient can distinguish that the hallucination is not real. EEG and EMG recordings during sleep and wakefulness show that narcoleptic patients fall asleep more frequently during the day, but they also awaken more frequently at night, so that they get about the same amount of sleep as normal individuals. However, they often enter into REM sleep very soon after sleep onset (short-onset REM periods [SOREMPs]), and during cataplexy attacks they show muscle atonia consistent with intrusion of a REM-like state into consciousness. On a multiple sleep latency test (MSLT), where the patient lies down in a quiet room five times during the course of the day at 2-hour intervals, narcoleptics typically fall asleep much faster than normal individuals (often in less than 5 minutes on repeated occasions) and show SOREMPs, which normal individuals rarely, if ever, experience.
There is a clear genetic predisposition to narcolepsy, as individuals with a firstdegree relative with the disorder are 40 times more likely to develop it themselves.38 However, there are clearly environmental factors involved, even among monozygotic twins; if one twin develops narcolepsy, the other will develop it only about 25% of the time. HLA allele DQB1*0602 is found in 88% to 98% of individuals with narcolepsy with cataplexy, but only in about 12% of white Americans and 38% of African Americans in the general population.
Scientists worked fruitlessly for decades to unravel the pathophysiology of this mysterious illness, until in 1999 two dramatic and simultaneous findings suddenly brought the problem into focus. The previous year, two groups of scientists, Masashi Yanagisawa and colleagues at the University of Texas Southwestern Medical School, and Greg Sutcliffe and coworkers at the Scripps Institute, had simultaneously identified a new pair of peptide neurotransmitters made by neurons in the lateral hypothalamus, which Yanagisawa called ‘‘orexins’’ (based on the pre-
(continued)
20
sumption of a role in feeding)67 and Sutcliffe called ‘‘hypocretins’’ (because it was a hypothalamic peptide with a sequence similar to secretin).68 Yanagisawa further showed that the type 1 orexin receptor had 10-fold specificity for orexin A, whereas the type 2 receptor was activated equally well by both orexins.69 The orexin neurons in the lateral hypothalamus were found to have wide-ranging projections
from the cerebral cortex to the spinal cord, much like the monoaminergic neurons in the brainstem.58,70
When Yanagisawa’s group prepared mice in which the orexin gene had been deleted, they initially found that the animals had normal sleep behavior during the day.70 However, when the mice were observed under infrared video monitoring during the night, they showed intermittent attacks of behavioral arrest during which they would suddenly fall over onto their side, twitch a bit, and lie still for a minute or two, before just as suddenly getting up and resuming their normal behaviors. EEG and EMG recordings demonstrated that these attacks have the appearance of cataplexy (sudden loss of muscle tone, EEG showing either an awake pattern or large amounts of theta activity typical of rodents during REM sleep). The animals also had short-onset REM periods when asleep, another hallmark of narcolepsy.
At the same time, Emmanuel Mignot had been working at Stanford for nearly a decade to determine the cause of genetically inherited canine narcolepsy. He fi- nally determined that the dogs had a genetic defect in the type 2 orexin receptor.71
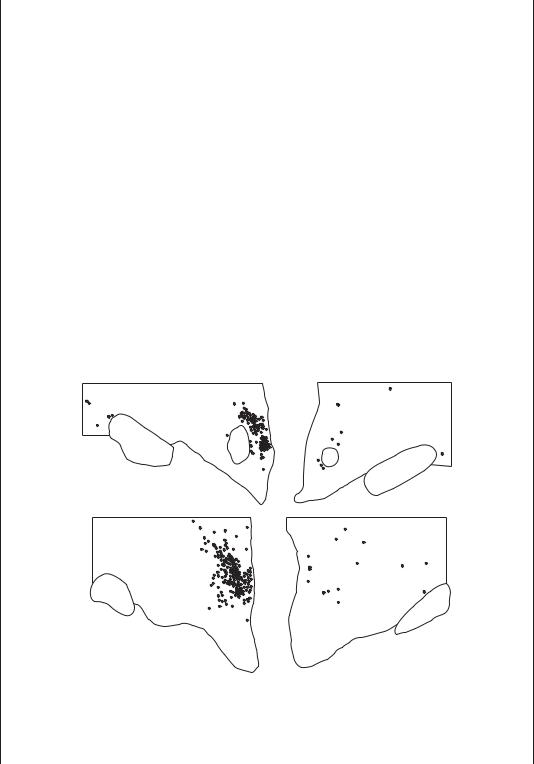
NORMAL |
NARCOLEPTIC |
112 |
9 |
237 |
MB |
MB |
15 |
|
Figure B1–4A. Narcolepsy is caused by loss of the orexin neurons in the posterior and lateral hypothalamus of the human brain. The panels plot the location of orexin neurons in the posterior hypothalamus in two subjects with normal brains on the left and two patients with narcolepsy on the right. There is typically about 90% loss of orexin neurons in patients who have narcolepsy with cataplexy. (From Thannickal, TC, Moore, RY, Nienhuis, R, et al. Reduced number of hypocretin neurons in human narcolepsy. Neuron 27, 469–474, 2000. By permission of Elsevier B.V.)
(continued)
21

22 Plum and Posner’s Diagnosis of Stupor and Coma
Box 1–4 Orexin and Narcolepsy (cont.)
The nearly simultaneous publication of the two results established firmly that narcolepsy could be produced in animals by impairment of orexin signaling.
Over the following year, it became clear that most humans with narcolepsy do not have a genetic defect either of the orexin gene or of its receptors, although a few cases with onset during infancy and particularly severe narcolepsy were found to be due to this cause.72 Instead, postmortem studies showed that narcoleptics with cataplexy lose about 90% of their orexin neurons, and that the spinal fluid levels of orexin often are very low.72–74 However, the nearby melanin-concentrating hormone neurons were not affected. This specificity suggested either an autoimmune or neurodegenerative cause of the orexin cell loss.
The presence of type 2 orexin receptors on histaminergic neurons, type 1 receptors in the locus coeruleus, and both types of orexin receptors on serotoninergic and other neurons in the pontine reticular formation75 suggests that one or more of these targets may be critical for regulating the transitions to REM sleep that are disrupted in patients with narcolepsy.
different pathways may fire independently under a variety of different conditions, modulating the functional capacities of cortical neurons during a wide range of behavioral states.
Behavioral State Switching
An important feature of the ascending arousal system is its interconnectivity: the cell groups that contribute to the system also maintain substantial connections with other components of the system. Another important property of the system is that nearly all of these components receive inputs from the ventrolateral preoptic nucleus.82–84 Ventrolateral preoptic neurons
contain the inhibitory transmitters GABA and galanin; they fire fastest during sleep.40,83,85 Le-
sions of the ventrolateral preoptic nucleus cause a state of profound insomnia in animals,86,87 and
such lesions undoubtedly accounted for the insomniac patients described by von Economo19 (see Box 1–1).
The ventrolateral preoptic neurons also receive extensive inhibitory inputs from many components of the ascending arousal system. This mutual inhibition between the ventrolateral preoptic nucleus and the ascending arousal system has interesting implications for the mechanisms of the natural switching from wakefulness to sleep over the course of the day, and from slow-wave to REM sleep over the course
of the night. Electrical engineers call a circuit in which the two sides inhibit each other a ‘‘flipflop’’ switch.84 Each side of a flip-flop circuit is self-reinforcing (i.e., when the neurons are firing, they inhibit neurons that would otherwise turn them off, and hence they are disinhibited by their own activity). As a result, firing by each side of the circuit tends to be self-perpetuating, and the circuit tends to spend nearly all of its time with either one side or the other in ascendancy, and very little time in transition. These sharp boundaries between wakefulness and sleep are a key feature of normal physiology, as it would be maladaptive for animals to walk around half-asleep or to spend long portions of their normal sleep cycle half-awake.
REM sleep is a stage of sleep in which the brain enters a very different state from the highvoltage slow waves that characterize NREM sleep. As indicated in Box 1–3, during REM sleep, the forebrain shows low-voltage, fast EEG activity similar to wakefulness, and the ascending cholinergic system is even more active than during a wakeful state. However, the ascending monoaminergic systems cease firing virtually completely during REM sleep,46–49 so that the increased thalamocortical transmission seen during REM sleep falls upon a cerebral cortex that lacks the priming to maintain a wakeful state. As a result, REM sleep is sometimes called paradoxical sleep because the cortex gives an EEG appearance of wakefulness, and yet the
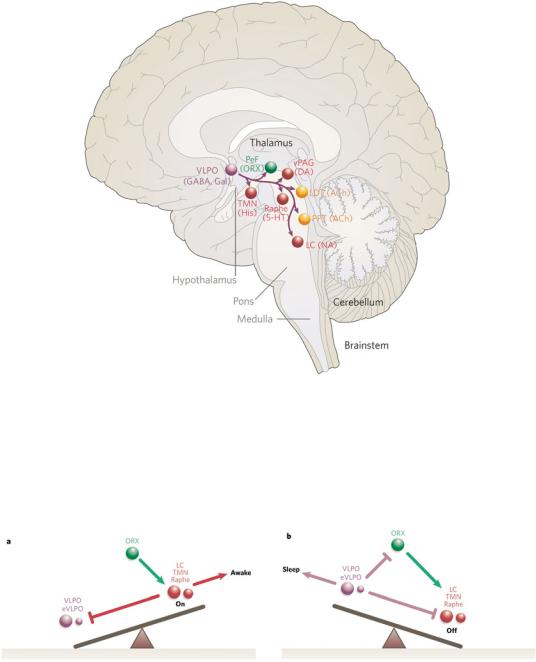
Figure 1–3. The ventrolateral preoptic nucleus (VLPO), shown in purple, inhibits the components of the ascending arousal system during sleep. VLPO neurons contain both gamma-aminobutyric acid (GABA) and an inhibitory peptide, galanin, and send axons to most of the cell groups that compose the ascending arousal system. This unique relationship allows the VLPO neurons effectively to turn off the arousal systems during sleep. Loss of VLPO neurons results in profound insomnia. 5- HT, serotonin; ACh, acetylcholine; DA, dopamine; Gal, ; His, histamine; LC, locus coeruleus; LDT, laterodorsal tegmental nuclei; NA, noradrenaline; ORX, orexin; PeF, ; PPT, pedunculopontine; TMN, tuberomammillary nucleus; vPAG, ventral periaqueductal gray matter. (From Saper, CB, Scammell, TE, Lu J. Hypothalamic regulation of sleep and circadian rhythms. Nature 437:1257–1263, 2005. By permission of Nature Publishing Group.)
Figure 1–4. A diagram of the flip-flop relationship between the ventrolateral preoptic nucleus (VLPO), which promotes sleep, and several monoaminergic cell groups that contribute to the arousal system, including the locus coeruleus (LC), the tuberomammillary nucleus (TMN), and raphe nuclei. During wakefulness (a), the orexin neurons (ORX) are active, stimulating the monoamine nuclei, which both cause arousal and inhibit the VLPO to prevent sleep. During sleep (b), the VLPO and extended VLPO (eVLPO) inhibit the monoamine groups and the orexin neurons, thus preventing arousal. This mutually inhibitory relationship ensures that transitions between wake and sleep are rapid and complete. (From Saper, CB, Scammell, TE, Lu J. Hypothalamic regulation of sleep and circadian rhythms. Nature 437:1257–1263, 2005. By permission of Nature Publishing Group.)
23
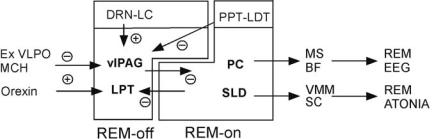
24 Plum and Posner’s Diagnosis of Stupor and Coma
Figure 1–5. The control elements for rapid eye movement (REM) sleep also form a flip-flop switch. Gamma-aminobutyric acid (GABA)-ergic REM-off neurons in the ventrolateral periaqueductal gray matter (vlPAG) and the lateral pontine tegmentum (LPT) inhibit the REM-on neurons in the sublaterodorsal (SLD) and the precoeruleus (PC) areas, whereas GABAergic SLD neurons inhibit the vlPAG and the LPT. This mutual inhibition forms a second flip-flop switch that regulates transitions into and out of REM sleep, which also are generally rapid and complete. Other modulatory systems, such as the extended ventrolateral preoptic nucleus (Ex VLPO) and the melanin-concentrating hormone (MCH) and orexin neurons in the hypothalamus, regulate REM sleep by their inputs to this switch. Similarly, the monoaminergic dorsal raphe nucleus (DRN) and locus coeruleus (LC) inhibit REM sleep by activating the REM-off neurons, and cholinergic neurons in the pedunculopontine (PPT) and laterodorsal tegmental nuclei (LDT) activate REM sleep by inhibiting neurons in the REMoff region. Neurons in the SLD cause motor atonia during REM sleep by excitatory inputs to inhibitory interneurons in the ventromedial medulla (VMM) and the spinal cord (SC), which inhibit alpha motor neurons. Neurons in the PC contact the medial septum (MS) and basal forebrain (BF), which drive the electroencephalogram (EEG) phenomena associated with REM sleep. (Modified from Lu, Sherman, Devor, et al.,53 by permission.)
individual is profoundly unresponsive to external stimuli.
A second flip-flop switch in the pons for switching from NREM to REM sleep (and back again) has recently been identified in the rostral pons. Many GABAergic neurons in the extended part of the ventrolateral preoptic nucleus are specifically active during REM sleep, suggesting that they inhibit a population of REM-off neurons.88 In addition, the orexin neurons in the lateral hypothalamus are excitatory, but their firing inhibits REM sleep, suggesting that they may activate REM-off neurons, as patients or animals with narcolepsy who lack orexin neurons transition into REM sleep exceptionally quickly.70,89 By searching for the intersection of these two pathways, a population of neurons was defined in the rostral pons, including the ventrolateral periaqueductal gray matter and the lateral pontine tegmentum at the level where they are adjacent to the dorsal raphe nucleus. These sites contain many GABAergic neurons, and lesions of this region increase REM sleep, confirming a REM-off influence.53 GABAergic neurons in the REM-off area innervate an adjacent region including the sublaterodorsal nucleus and pre-coeruleus region that contain REM-active neurons. This REM-on region contains two types of neurons. GABAergic neurons, mainly in the sublaterodorsal nucleus, project back to the REM-off area. This produces a flip-
flop switch relationship accounting for the tendency for transitions into and out of REM sleep to be relatively abrupt. A second population of neurons is glutamatergic. Glutamatergic REMon neurons in the sublaterodorsal nucleus project to the brainstem and spinal cord, where they are thought to be responsible for the motor manifestations of REM sleep, including atonia and perhaps the rapid eye movements that are the hallmarks of the state. Glutamatergic REMon neurons in the coeruleus region target the basal forebrain where they appear to be critical for maintaining EEG phenomena associated with REM sleep.
Cholinergic and monoaminergic influences may have a modulatory effect on REM sleep by playing upon this flip-flop switch mechanism. Although lesions of these systems do not have a major effect on REM sleep, overactivity may have quite dramatic effects. For example, injections of cholinomimetic agents into the region containing the REM switch can trigger prolonged bouts of a REM-like state in animals.90 Whether this is due to activating REM-on neurons or inhibiting REM-off neurons (or both) is not known. On the other hand, patients who take antidepressants that are either serotonin or norepinephrine reuptake inhibitors (or both) have very little REM sleep. This effect may be due to the excess monoamines activating the REM-off neurons or inhibiting the REM-on
Pathophysiology of Signs and Symptoms of Coma |
25 |
neurons (or both) and thereby locking the individual out of REM sleep.70,89
Relationship of Coma to Sleep
Because the brain enters a state of quiescence during sleep on a daily basis, it is natural to wonder whether coma may not be a pathologic entrance into the sleep state. In fact, both impaired states of consciousness and NREM sleep are characterized by EEG patterns that include increased amounts of high-voltage slow waves. Both conditions are due, ultimately, to lack of activity by the ascending arousal system. However, in sleep, the lack of activity is due to an intrinsically regulated inhibition of the arousal system, whereas in coma the impairment of the arousal system is due either to damage to the arousal system or to diffuse dysfunction of its diencephalic or forebrain targets.
Because sleep is a regulated state, it has several characteristics that distinguish it from coma. A key feature of sleep is that the subject can be aroused from it to wakefulness. Patients who are obtunded may be aroused briefly, but they require continuous stimulation to maintain a wakeful state, and comatose patients may not be arousable at all. In addition, sleeping subjects undergo a variety of postural adjustments, including yawning, stretching, and turning, which are not seen in patients with pathologic impairment of level of consciousness.
The most important difference, however, is the lack of cycling between NREM and REM sleep in patients in coma. Sleeping subjects undergo a characteristic pattern of waxing and waning depth of NREM sleep during the night, punctuated by bouts of REM sleep, usually beginning when the NREM sleep reaches its lightest phase. The monotonic high-voltage slow waves in the EEG of the comatose patient indicate that although coma may share with NREM sleep the property of a low level of activity in the ascending arousal systems, it is a fundamentally different and pathologic state.
The Cerebral Hemispheres
and Conscious Behavior
The cerebral cortex acts like a massively parallel processor that breaks down the components of
sensory experience into a wide array of abstractions that are analyzed independently and in parallel during normal conscious experience.42 This organizational scheme predicts many of the properties of consciousness, and it sheds light on how these many parallel streams of cortical activity are reassimilated into a single conscious state.
The cerebral neocortex of mammals, from rodents to humans, consists of a sheet of neurons divided into six layers. Inputs from the thalamic relay nuclei arrive mainly in layer IV, which consists of small granule cells. Inputs from other cortical areas arrive into layers II, III, and V. Layers II and III consist of smallto mediumsized pyramidal cells, arrayed with their apical dendrites pointing toward the cortical surface. Layer V contains much larger pyramidal cells, also in the same orientation. The apical dendrites of the pyramidal cells in layers II, III, and V receive afferents from thalamic and cortical axons that course through layer I parallel to the cortical surface. Layer VI comprises a varied collection of neurons of different shapes and sizes (the polymorph layer). Layer III provides most projections to other cortical areas, whereas layer V provides long-range projections to the brainstem and spinal cord. The deep part of layer V projects to the striatum. Layer VI provides the reciprocal output from the cortex back to the thalamus.91
It has been known since the 1960s that the neurons in successive layers along a line drawn through the cerebral cortex perpendicular to
the pial surface all tend to be concerned with similar sensory or motor processes.92,93 These
neurons form columns, of about 0.3 to 0.5 mm in width, in which the nerve cells share incoming signals in a vertically integrated manner. Recordings of neurons in each successive layer of a column of visual cortex, for example, all respond to bars of light in a particular orientation in a particular part of the visual field. Columns of neurons send information to one another and to higher order association areas via projection cells in layer III and, to a lesser extent, layer V.94 In this way, columns of neurons are able to extract progressively more complex and abstract information from an incoming sensory stimulus. For example, neurons in a primary visual cortical area may be primarily concernedwithsimple lines,edges,andcorners, but by integrating their inputs, a neuron in a higher order visual association area may