

5. Основы химической кинетики и катализа
Законы термодинамики не дают ответа о скорости реакции, а наука, изучающая скорость реакций и их механизм называется химической кинетикой,
Первые систематические исследования скоростей химических реакций были выполнены Н.А.Меншуткиным в 70-х годах прошлого столетия. В 1884 году Я.Вант-Гофф сформулировал в общей форме кинетические закономерности протекания простых − моно- би- тримолеку-лярных реакций.
Толчком к дальнейшему развитию химической кинетики послужило установление Аррениусом (1889 г.) роли в химической реакции активных молекул и зависимости скорости простых реакций от температуры. Затем с использованием законов и приемов статистической физики и квантовой механики была создана теория активированного комплекса.
Усилиями различных ученых была установлена важная роль в кинетике сложных реакций промежуточных веществ. Изучение промежуточных веществ было стимулировано открытием обычных и разветвленных цепных реакций.
Было установлено, что промежуточные вещества представляют собой активные частицы − свободные атомы, радикалы, ионы, обладающие высокой реакционной способностью.
Скорость химической реакции в замкнутой системе определяется экспериментально по изменению во времени концентраций исходных, промежуточных веществ или продуктов реакции, Зависимость концентраций этих веществ от времени обычно представляется кривыми, которые называют кинетическими кривыми.
С помощью специальных приемов математической обработки из кинетических кривых получают кинетические параметры реакции − константу скорости, порядок реакции, энергию активации.
Скорость химической реакции зависит от температуры, Зависимость константы реакции от абсолютной температуры устанавливает уравнение Аррениуса:
К = А∙е-Е/RT, (5.1)
где: А и Е – соответственно, предэкспоненциальный множитель и энергия активации реакции.
Численные значения А и Е определяют графически, соответственно, по начальной ординате и углу наклона прямой линии, представляющей опытные данные в координатах lnК − 1/Т.
lnК
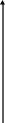







1/Т
Порядок реакции. Все химические реакции формально делятся на реакции первого порядка, второго и т. д. По тому, сколько молекул участвует одновременно в элементарном акте реакции, различают истинный порядок реакции: молекулярный, бимолекулярный и т. д.
Формально порядок реакции по данному веществу – это показатель степени концентрации этого вещества в кинетическом уравнении:
![]() (5.2)
(5.2)
где: V - скорость реакции,
nА - порядок реакции по веществу А;
К - константа скорости реакции (равна скорости реакции V при
концентрации реагентов равной 1).
Порядок реакции зависит от механизма реакции и может изменяться с температурой и давлением.
Для элементарных реакций порядок − это целочисленная величина, для сложных реакций он может быть дробным, а также отрицательным числом. Порядок реакции, как правило, не превышает 3. Порядок реакции определяется экспериментально, обычно по зависимости скорости реакции от концентрации данного вещества при постоянной концентрации всех остальных веществ. Порядок реакции позволяет судить о механизме реакции.
Энергия активации − это средняя избыточная энергия Е, которой должны обладать реагирующие частицы, чтобы преодолеть потенциальный барьер, разделяющий исходное и конечное состояние системы. Энергию активации обычно определяют по зависимости константы скорости реакции К от абсолютной температуры Т. Согласно уравнению Аррениуса тангенс угла наклона прямой в координатах lnК − 1/Т равен Е/4,575.
Различают истинную и эффективную (наблюдаемую) энергию активации в зависимости от того, истинную или эффективную константу скорости реакции определяют в эксперименте.
Для химически насыщенных молекул (молекулярные реакции) значение истинной энергии активации для элементарных реакций составляет 80-240 кДж/моль.
Для радикальных реакций она равна от нескольких до 60 кДж/моль, для ионных реакций и рекомбинации радикалов энергия активации вообще может не требоваться.
Механизм реакций − это совокупность составляющих реакцию элементарных процессов. Различают простые и сложные реакции. В механизм простой реакции входят:
1. физические процессы передачи энергии, которые лежат в основе активации и дезактивации реагирующих частиц;
2. собственно химическое превращение, т.е. перестройка химических связей.
Пример:
Мономолекулярная реакция превращения вещества А в В, т.е.
А → В может состоять из следующих элементарных процессов:
А + М → А*+ М (5.3)
А* + М → В + М (5.4)
где: А* - активная молекула;
М - любая частица в реагирующей системе, которая передает свою
кинетическую энергию молекуле А.
Сложные реакции состоят из нескольких элементарных стадий образования и превращения промежуточных веществ, которыми могут быть как молекулы, так и свободные атомы, радикалы, ионы, электроны, возбужденные частицы.
Установление механизма сложных реакций является очень сложной задачей. Эта задача при исследованиях разбивается на два этапа.
1 этап. Определение последовательности элементарных стадий
сложной реакции и соотношения их констант скорости;
2 этап. Исследование динамики каждой из элементарных стадий.
При исследовании первого этапа используют как прямые, так и косвенные методы.
Прямое исследование реакций включает идентификацию промежуточных веществ и измерение их концентраций с применением различных физико-химических методов.
При косвенном кинетическом исследовании на основании предполагаемого механизма составляют систему дифференциальных уравнений, включающую уравнения каждой из элементарных стадий. В простых случаях удается получить уравнение для суммарной скорости реакции. Сопоставляя решение этого уравнения с наблюдаемыми на опыте зависимостями скорости реакции от времени, судят о правильности (адекватности) предполагаемого механизма.
Описание предполагаемого механизма требует хорошего знания законов строения и реакционной способности органических соединений, законов химической термодинамики и катализа.
Для решения задачи на втором этапе применяют как экспериментальные, так и теоретические методы (квантовые химические расчеты).
Катализ - это изменение скорости или возбуждение химической реакции веществами, которые участвуют в реакции, но не входят в состав конечных продуктов.
Различают положительный катализ − ускорение реакции (катализаторы) и отрицательный катализ − замедление реакции (ингибиторы). Обычно термин катализ относится к ускорению реакции.
Катализатором называют вещество, многократно вступающее в промежуточное химическое взаимодействие с реагентами, не участвующее в стехиометрическом уравнении реакции, не изменяющее термодинамическое равновесие, но увеличивающее скорость его достижения, т.е. скорость реакции.
То, что катализатор не участвует в стехиометрическом уравнении реакции, не означает неизменности его состава и свойств, Так, под влиянием реагентов, примесей к реагентам, основных и побочных продуктов реакции, температуры катализатор всегда претерпевает физико-химические изменения.
Иногда, в результате взаимодействия с одним из продуктов реакции свойства катализатора изменяются очень быстро, но при удалении этого продукта он восстанавливает свои первоначальные свойства − регенирируется.
Катализатор не влияет на термодинамическое равновесие реакции, если можно пренебречь изменением энергии Гиббса в результате образования стабильных продуктов взаимодействия реагентов или продуктов реакции с катализатором. Обычно изменением ∆G реакции под влиянием катализатора можно пренебречь, так как образование продуктов побочной реакции с катализатором очень мало относительно образования основных продуктов реакции.
Равенство изменения энергии Гиббса при реакции в присутствии катализатора и без него означает, что катализатор в одинаковой степени ускоряет и прямую и обратную реакции. Если реакция проводится в условиях, близких к равновесным, то данный катализатор можно применять для ускорения как прямой, так и обратной реакции (например: гидрирование и дегидрирование).
Многие реакции чисто термически, без использования катализатора, не осуществляются вообще (например: изомеризация парафиновых углеводородов), Однако, термодинамически эти реакции возможны, и применение катализатора позволяет осуществить их. Катализатор воздействует на реакцию в результате промежуточного химического взаимодействия с реагирующими веществами, которое открывает новый, невозможный без катализатора путь реакции, состоящий, как правило, из большего числа элементарных стадий, чем в отсутствие катализатора.
Влияние энергии активации
на протекание химической реакции
Скорости некаталитических и каталитических реакций связаны с энергией активации и в газовой фазе, если протекает мономолекулярная реакция А → В равны:
![]() (5.5)
(5.5)
Для мономолекулярных реакций:
Ко(терм.) ≈ 1013 с-1, но не может быть < этого значения;
Ко(кат.) ≈ 105 с-1, но не может быть > этого значения и равно удельному числу соударений молекул с поверхностью (S) катализатора.
Чтобы каталитическая реакция осуществлялась, энергия активации ее должна быть ниже, чем некаталитической. Следовательно, катализ обусловлен тем, что энергия активации каталитической реакции всегда меньше энергии активации той же реакции, но протекающей без катализатора, т.е. термической. Почему же Eкaт. < Eтерм.? Потому, что каталитическая реакция более сложная и состоит из большего числа элементарных стадий, a Eкат. любой стадии всегда меньше Eтерм.. В этом и заключается основное условие катализа.
Зависимость энергии активации от пути реакции можно рассмотреть графически:







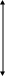










 Еа
Еа
 Е1
Е1
А+К
Е2
В
В+К
АК


 некаталитическая
реакция каталитическая реакция
некаталитическая
реакция каталитическая реакция
п у т ь р е а к ц и и
где: А – исходный реагент;
В – конечный продукт;
К – катализатор;
Е – энергия активации;
∆Q – тепловой эффект.
Каталитическое превращение может осуществляться по стадийному механизму, когда одна некаталитическая реакция типа
А + В → С + D (5.6)
заменяется совокупностью нескольких стадий последовательного взаимодействия реагентов с катализатором с образованием на каждой стадии активированного комплекса:
А + К → (АК)* → АК (5.7)
АК + В → (АВК)* → С + D + К (5.8)
или по слитному (ассоциативному, синхронному) механизму, включающему одновременное взаимодействие всех реагентов с катализатором и образование одного активированного комплекса:
А + В + К → (АВК)* → С + D + К (5.9)
Гомогенный и гетерогенный катализ
Существует гомогенный и гетерогенный катализ.
В зависимости от того, находится ли катализатор в той же фазе, что и реагирующие вещества, или образуют самостоятельную фазу, говорят о гомогенном или гетерогенном катализе.
Если реагенты и катализатор находятся в одной фазе (газовая смесь или раствор), то осуществляется гомогенный катализ.
При гетерогенном катализе катализатор - твердое вещество, а реагенты - жидкие или газообразные вещества. Реакция в этом случае протекает на поверхности катализатора.
Гетерогенный катализ распространен в нефтеперерабатывающей промышленности значительно больше, чем гомогенный.
Современная теория катализа считает, что при гомогенном катализе осуществляется новый путь реакции, состоящий из нескольких стадий, в ходе которых образуются и разлагаются неустойчивые про-межуточные соединения реагентов с катализатором. Ускорение реакции объясняется тем, что скорость каждой стадии больше, чем скорость некаталитической реакции.
Адсорбционная теория гетерогенного катализа ускорение реакции объясняет деформирующим (разрыхляющим) действием катализатора на химические связи в реагирующих молекулах.
Электронные теории гетерогенного катализа каталитическую активность металлов и полупроводников объясняют наличием электронов проводимости, которые могут принимать участие в окислительно-восстановительных превращениях на поверхности катализатора.
Основные стадии гетерогенного каталитического процесса. Реакция газообразных реагентов на твердом катализаторе складывается из следующих стадий.
1. Диффузия реагентов из газового потока к внешней поверхности частиц катализатора (внешняя диффузия).
2. Диффузия реагентов в поры катализатора (внутренняя диффузия).
3. Адсорбция реагентов на поверхности катализатора.
4. Химическая реакция на поверхности катализатора.
5. Десорбция продуктов реакции.
6. Диффузия продуктов реакции в порах катализатора.
7. Диффузия продуктов реакции от внешней поверхности частиц катализатора в газовый поток,
При этом, стадии (2) - диффузия реагентов в поры катализатора и (6) - диффузия продуктов реакции в порах катализатора осуществляются только на пористом катализаторе.
При установившемся режиме скорости всех стадий равны. Обычно одна из стадий является лимитирующей, т.е. скорость которой предельна и не может повыситься при повышении скорости остальных стадий. В зависимости от того, какая из стадий является лимитирующей, различают следующие области протекания реакции.
1. Внешнедиффузионная область. Скорость реакции, протекающей во внешнедиффузионной области, практически не зависит от температуры, а зависит от скорости потока и размера частиц катализатора. Поверхность пор катализатора и его активность на скорость реакции в этом случае не влияют.
Если основная и побочная реакции имеют одинаковые порядки и побочная реакция имеет значительно большую энергию активации, чем основная, то при протекании реакции во внешнедиффузионной области побочная реакция может сильно ускориться относительно основной.
2. Внутридиффузионная область. Протекание реакций в этой области обусловлено наличием пор в катализаторе.
Если реакция протекает медленно относительно внешней диффузии, то молекулы реагентов диффундируют в поры катализатора. По мере протекания реакции по длине пор катализатора концентрация реагентов уменьшается.
Если реакция протекает достаточно быстро, то уже недалеко от устья поры концентрация реагентов равна нулю, и реакция проходит только на части поверхности поры. Такой механизм протекания реакции называется внутридиффузионным.
В данном случае необходимо учитывать соотношение между диаметром пор катализатора и их длиной. В противном случае будет отсутствовать процесс столкновения молекул реагентов об стенки катализатора и между собой.
3. Кинетическая область. Если реакция на поверхности быстрее диффузии в поры, но медленнее внешней диффузии, то реакция также протекает на поверхности катализатора независимо от диффузии. Это случай протекания реакции во внешней кинетической области.
Если скорость процесса определяется механизмом реакции на всей внутренней и внешней поверхности катализатора, то процесс проходит во внутренней кинетический области.
В первом случае скорость реакции пропорциональна внешней поверхности частиц катализатора, во втором - его удельной поверхности.
Скорость реакции в кинетической области может определяться:
скоростью адсорбции;
скоростью реакции на поверхности адсорбированных молекул;
скоростью реакции молекул, соударяющихся из газовой фазы с адсорбированными молекулами;
скоростью десорбции.
Классификация каталитических
реакций и катализаторов
Все катализаторы, применяемые в нефтепереработке и нефтехимии, по характеру взаимодействия катализатора с реагирующими веществами и по типу промежуточных продуктов разделяют на кислотно-основные и окислительно-восстановительные.
В кислотно-основных реакциях промежуточными активными частицами являются ионы, и катализатор инициирует их образование в результате передачи протонов от катализатора к реагенту или от реагента к катализатору. Катализаторами в этом случае являются кислоты (кислотный катализ) и основания (основной катализ).
В окислительно-восстановительных реакциях промежуточными активными частицами являются радикалоподобные нейтральные образования и каталитическое воздействие связано с переходом электрона от молекулы катализатора к молекуле реагента и наоборот.
Кислотно-основные и окислительно-восстановительные функции катализаторов могут быть совмещены и тогда катализаторы имеют двойную активность, т.е. называются бифункциональными катализаторами.
Многие промышленные катализаторы являются бифункциональными, так как окислительно-восстановительный катализатор наносят на носитель, являющийся кислотным катализатором.
В нефтеперерабатывающей промышленности
1. К кислотному катализу можно отнести:
- каталитический крекинг;
- алкилирование изобутана бутиленом;
- полимеризацию олефинов.
2. К окислительно-восстановительному:
- процессы гидрогенизации и дегидрогенизации.
3. Бифункциональный катализ широко применяется в процессах:
- риформинга;
- гидрокрекинга;
- изомеризации парафиновых углеводородов.
Катализ основаниями в нефтеперерабатывающей промышленности не применяется.
Одна и та же реакция может происходить в присутствии различных катализаторов.
Скорость данной реакции в присутствии различных катализаторов характеризует их активность относительно этой реакции.
В подавляющем большинстве случаев в присутствии данного катализатора помимо основной реакции протекает еще ряд параллельных и последовательных реакций, и исходные вещества превращаются в смесь различных продуктов.
Доля прореагировавших исходных веществ, превращаемая в присутствии данного катализатора в желаемые продукты, характеризует его селективность.
Важнейшим свойством катализатора является его способность сохранять активность во времени, характеризуемая стабильностью.
При гомогенном катализе жидкий катализатор дезактивируется в процессе работы в результате накопления в нем продуктов, снижающих его концентрацию. Это происходит в результате физического и химического растворения в катализаторе примесей к сырью или побочных продуктов каталитической реакции.
Значительно многообразнее причины снижения активности твердых катализаторов (гетерогенный катализ). Катализаторы могут претерпевать как физические, так и химические изменения.
Физическим изменениям подвергаются макро- и микро- структуры катализатора. Под температурным воздействием происходит рекристаллизация металлов, приводящая к уменьшению удельной поверхности катализатора или числа активных центров на единице его поверхности. Механические и термические воздействия на катализатор приводят к постепенному разрушению его частиц. Во избежание протекания этих процессов в состав катализатора вводят специальные добавки.
Химические изменения катализатора в процессе его вызываются хемосорбцией на его поверхности примесей к сырью или продуктов разложения этих примесей. В результате очень быстро может снизиться активность катализатора. Такое быстрое снижение активности называется отравлением катализатора, а примеси – каталитическими ядами. Если активность катализатора после удаления из сырья примесей (каталитических ядов) через некоторое время восстанавливается, то отравление называется обратимым, и наоборот.
Обратимость или необратимость отравления связана с прочностью хемосорбции яда на поверхности катализатора. Если сырье и реагент могут постепенно вытеснить яд с поверхности катализатора, то отравление обратимо.
При каталитической переработке углеводородов на поверхности катализатора накапливаются высокомолекулярные, обедненные водородом продукты, называемые коксом. Отложения кокса, покрывая активную поверхность катализатора, прекращают доступ к ней молекул сырья. Удаление коксовых отложений с поверхности катализатора путем газификации кислородом, двуокисью углерода или водяным паром приводит к восстановлению активности катализатора.
В состав современных катализаторов вводят редкие и редкоземельные металлы. Присутствие их в составе катализатора предотвращает рекристаллизацию металлов, выполняющих гидрирующе-дегидрирующую функцию, что стабилизирует активность катализатора. Кроме того, повышается стабильность катализатора к закоксовыванию.
На сохранение активности катализатора в процессе его эксплуатации сильно влияет содержание гетероатомных соединений в исходном сырье. Поэтому сырье подвергается предварительной гидроочистке. Кроме серы на активность катализатора влияют соединения тяжелых металлов и их окислов.
Если отравление катализатора коксом и серой является обратимым, то отравление металлами – необратимым.
Взаимосвязь скорости реакции
и времени ее протекания
Для реакции первого порядка константа скорости реакции определяется по уравнению:
![]() или
или
![]() (5.10)
(5.10)
где: τ – время реакции, с;
а – начальная концентрация исходного вещества;
х – уменьшение концентрации исходного вещества с момента
начала реакции до данного момента;
а - х – концентрация исходного вещества к данному моменту
времени.
Период полураспада для реакции первого порядка выражается уравнением:
![]() (5.11)
(5.11)
и не зависит от начальных концентраций исходных веществ.
Для реакции второго порядка:
![]() или
или
![]() (5.12)
(5.12)
где: а и b – начальные концентрации исходных веществ;
х – уменьшение концентрации начальных веществ с начала
реакции до данного момента;
(а – х) и (b – х) – концентрации исходных веществ к данному моменту
времени.
В случае, когда а = b:
![]() (5.13)
(5.13)
Период полураспада для реакции второго порядка:
![]() (5.14)
(5.14)
Для реакции третьего порядка при условии а = b = с:
![]() (5.15)
(5.15)
Существуют также реакции, в которых скорость процесса не зависит от концентрации, т.к. она определяется некоторыми другими факторами, например, скоростью диффузии при поверхностных реакциях. И тогда, реакцию считают реакцией нулевого порядка, а константу скорости определяют по уравнению:
![]() (5.16)
(5.16)
Для мономолекулярной обратимой реакции
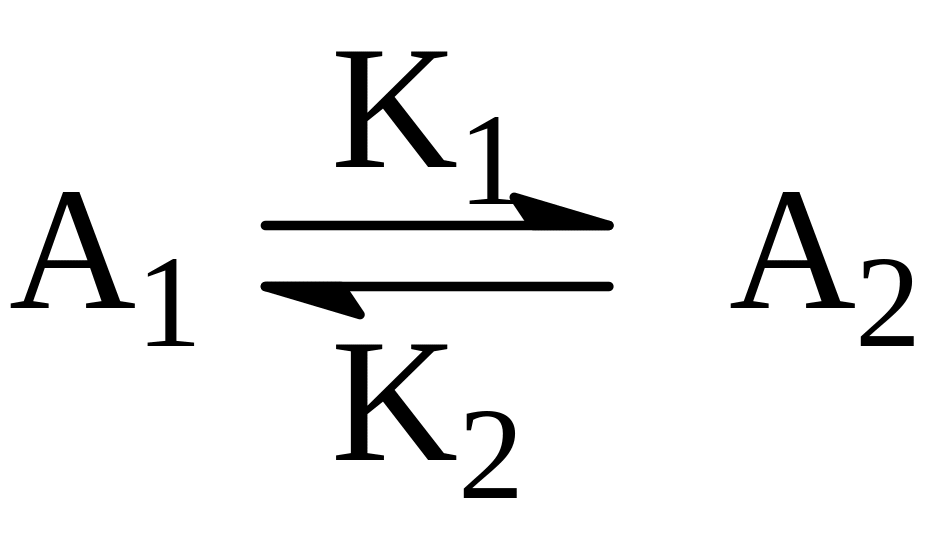
действительная скорость реакции в данный момент равна разности скоростей прямой и обратной реакции:
![]() (5.17)
(5.17)
где: К1 и К2 – константы скорости прямой и обратной реакций.
Зависимость скорости реакции от температуры
Если значения
констант скорости реакции
![]() и
и
![]() для
двух температур Т1
и Т2
известны, то по уравнению:
для
двух температур Т1
и Т2
известны, то по уравнению:
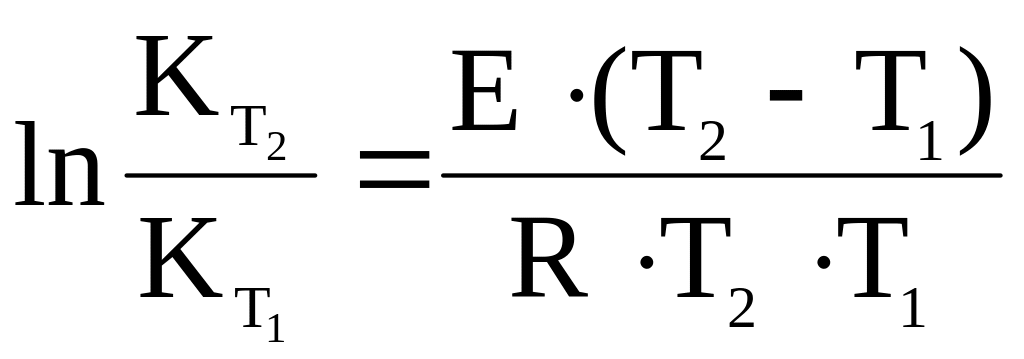 или
или
 (5.18)
(5.18)
можно определить значение энергии активации Еа для данной реакции:
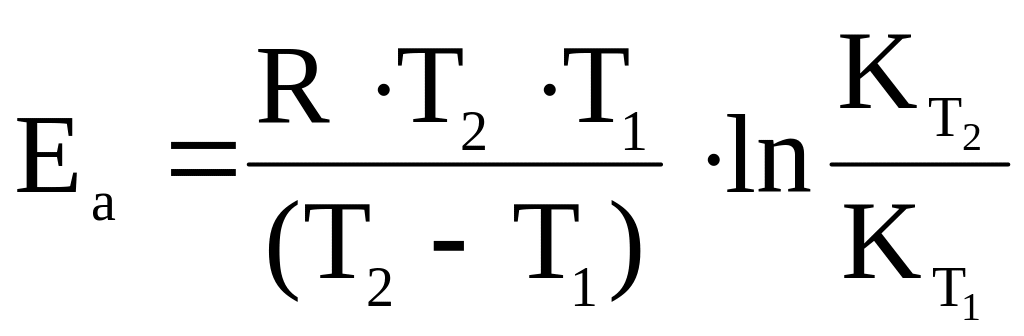 или
или
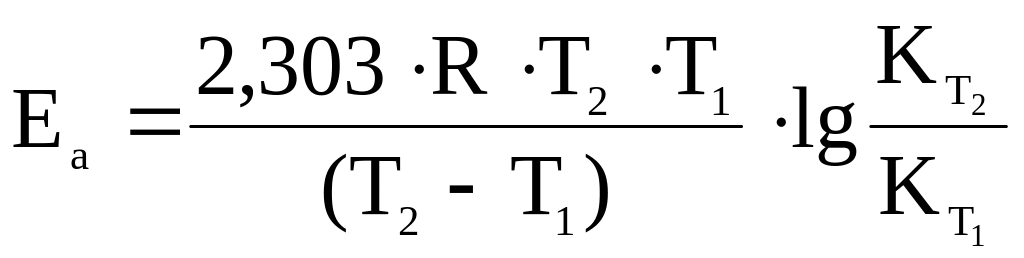 (5.19)
(5.19)
При решении практических задач необходимо знание таких понятий, как "температурный коэффициент реакции" и "температурный градиент реакции".
Величина, показывающая, во сколько раз изменится константа скорости реакции при изменении температуры на 10о, называется "температурным
показывающая, на сколько градусов нужно изменить коэффициентом реакции" (γ).
Величина, температуру реакции, чтобы скорость ее изменилась в два раза, называется "температурным градиентом реакции" (α).
Зависимость энергии активации
от теплового эффекта епты
Энергия активации реакций радикалов − замещения, отрыва, распада, присоединения по кратной связи, изомеризации и алкилирования зависит от теплового эффекта и определяется уравнениями Поляни-Семенова:
Еа = 48,2 – 0,25Q (кДж/моль) - для экзотермических реакций (5.20)
Еа = 48,2 + 0,75Q (кДж/моль) - для эндотермических реакций (5.21)
Методы определения порядка реакции
1. Метод подстановки.
Метод заключается в определении, какое уравнение (1-го; 2-го; 3-го порядка) при подстановке в него экспериментальных данных дает при решении постоянное значение константы скорости реакции. Именно это уравнение и определяет порядок исследуемой реакции.
2. Графический метод.
Метод заключается в построении графика, выражающего зависимость различных функций концентрации от времени:
lnC = f (τ) - 1 порядок; (5.22)
1/С = f (τ) - 2 порядок; (5.23)
1/С2 = f (τ) - 3 порядок, (5.24)
а затем, в определении, для какой из них зависимость выражается прямой линений. именно эта функция и будет определять порядок реакции.
3. Метод определения периода полураспада.
Метод основан на определении промежутка времени, в течение которого концентрации или количества исходных веществ уменьшаются ровно в два раза:
- если τ1/2 не зависит от концентрации, то реакция первого порядка;
- если τ1/2 обратно пропорционален начальной концентрации в первой степени, то реакция второго порядка;
- если τ1/2 обратно пропорционален начальной концентрации во второй степени, то реакция третьего порядка.
Примеры решения задач Задача 1. Для реакции 1-го порядка период полупревращения при температуре 600оС равен 2 сек. Определите константу скорости реакции в этих условиях. Какое время необходимо, чтобы реакция прошла на 75%?
Решение: Используя уравнение для определения периода полураспада (5.11) реакции первого порядка находим:
τ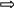 1/2
= 0,693/К К = 0,693/2 = 0,3465 с-1,
1/2
= 0,693/К К = 0,693/2 = 0,3465 с-1,
затем по уравнению (5.10) реакции первого порядка, зная значение константы скорости, вычисляем время, за которое реакция пройдет на 75%:
![]() сек.
сек.
Задача 2. Определите энергию активации реакции при температуре 427оС, если температурный градиент ее α = 8оС.
Решение: Для расчета используем уравнение (5.19) зависимости энергии активации от температуры:
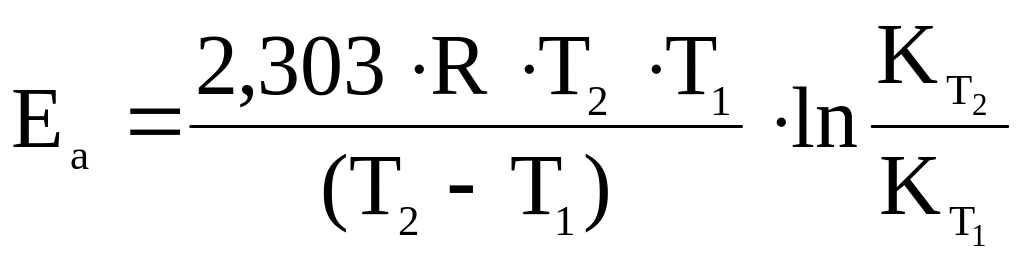 ,
предварительно вычислив
,
предварительно вычислив
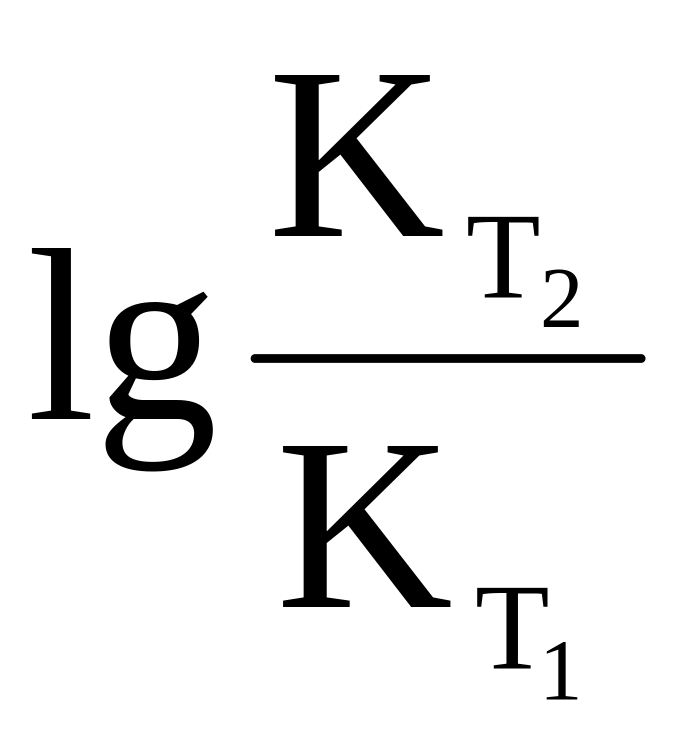 .
.
По определению понятия "температурный градиент реакции":
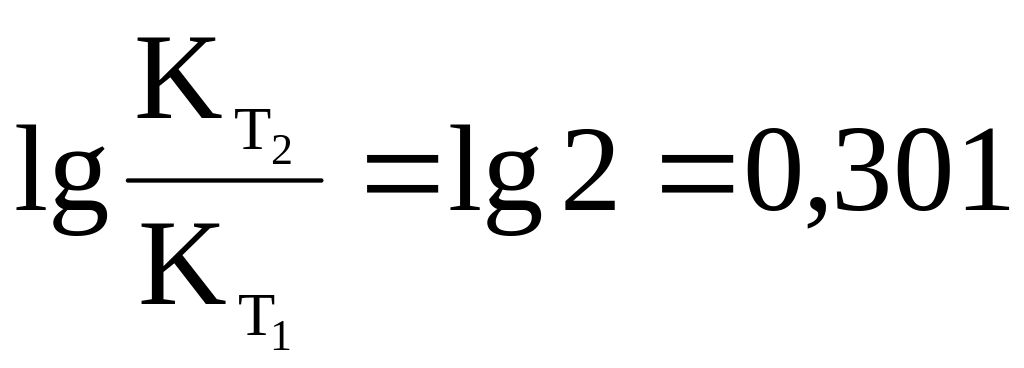 ,
тогда
,
тогда
![]() Дж/моль ≈ 356,4
кДж/моль: Задача
3.
Реакция
разложения этана: C2H6
↔ C2H4
+ Н2
при температуре 820оС
протекает на 75% за 1,1 сек. За какое время
она достигнет этой же глубины при
температуре 860оС,
если энергия активации этой реакции Еа
= 68Ю1 кДж/моль (порядок реакции – первый)?
Дж/моль ≈ 356,4
кДж/моль: Задача
3.
Реакция
разложения этана: C2H6
↔ C2H4
+ Н2
при температуре 820оС
протекает на 75% за 1,1 сек. За какое время
она достигнет этой же глубины при
температуре 860оС,
если энергия активации этой реакции Еа
= 68Ю1 кДж/моль (порядок реакции – первый)?
Решение:
По уравнению (5.10) реакции первого порядка находим константу скорости:
![]() =
=
![]() с-1
с-1
Затем вычисляем логарифм отношения скоростей реакции при двух температурах (820 оС и 860 оС):
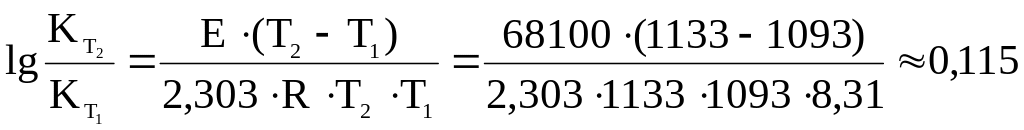
Отношение скоростей реакции будет равно:

![]() с-1
с-1
Тогда, время протекания реакции при температуре 860 оС будет равно:
![]() сек
сек