

3 курс / Фармакология / Диссертация_Ромодановский_Д_П_Разработка_требований_для_оценки_фармакокинетики
.pdfэкономического союза», подход к исследованиям биоэквивалентности препаратов с УТД
полностью гармонизирован с Европейским руководством EMA [35, 78].
Таблица 12.
Список лекарственных препаратов с узким терапевтическим индексом.
Список FDA, 1995 г. (в настоящее время не |
аминофилин, |
карбамазепин, |
клиндамицин, |
||||
применяется) |
клонидин, |
дифилин, |
дизопирамид, |
этинил- |
|||
|
эстрадиол/прогестин, |
|
гуанетидин, |
изоэтарин, |
|||
|
изопротеренол, лития карбонат, метапротеренол, |
||||||
|
миноксидил, окстрифилин, фенитоин, празозин, |
||||||
|
примидон, прокаинамид, хинидин, теофиллин, |
||||||
|
вальпроевая кислота, дивалпроекс натрия, варфарин. |
||||||
Список «Health Canada» [ (CanadaBE)] |
циклоспорин, |
дигоксин, |
флекаинид, |
литий, |
|||
|
фенитоин, сиролимус, теофиллин, варфарин. |
|
|||||
Список NIHS [ (JapanBE)] |
априндин, |
кармазепин, |
клиндамицин, |
||||
|
клоназепам, клонидин, циклоспорин, дигитоксин, |
||||||
|
дигоксин, |
дизопирамид, |
этинил-эстрадиол, |
||||
|
этосуксимид, гуанетидин, изопреналин, литий, |
||||||
|
метотрексат, фенобарбитал, фенитоин, празосин, |
||||||
|
примидон, |
прокаинамид, |
|
хинидин, |
|||
|
гипогликемические |
|
средства |
|
группы |
||
|
сульфонилмочевины (ацетогексамид, глибенкламид, |
||||||
|
гликлазид, гликопирамид, толазамид, толбутамид), |
||||||
|
такролимус, |
производные |
|
теофиллина |
|||
|
(аминофиллин, теофиллинат холина, дипрофиллин, |
||||||
|
проксифиллин, теофиллин), вальпроевая кислота, |
||||||
|
варфарин, зонисамид, глибузол. |
|
|
|
|||
Примечания: FDA – Администрация по пищевым продуктам и лекарственным средствам Соединенных Штатов Америки; Health Canada - Канадский регулятор обращения лекарственных средств; NIHS – Японский Национальный институт медицинских наук.
Таким образом, ЛП с УТД - это препараты, у которых терапевтический диапазон - разница между терапевтической и токсической сывороточными концентрациями лекарственных средств,
сужен относительно такового для большинства препаратов, что говорит о том, что такие препараты в терапевтических дозах могут вызывать серьезные нежелательные реакции.
3.1.2.2 Дизайн исследования и субъекты исследования
Для ЛП с УТД можно рекомендовать два основных вида дизайна исследований БЭ:
простой перекрестный с двумя периодами в двух последовательностях («2х2х2») и полностью повторный с четырьмя периодами в двух последовательностях («2х2х4»), как описано выше. Как правило, в исследование можно включать здоровых добровольцев.
В случае небезопасности многократного применения исследуемого лекарственного средства у здоровых добровольцев возможно включение сопоставимой по критериям включения/невключения популяции пациентов или использовать параллельный дизайн исследования, т.к. в данном случае отсутствует необходимость приема нескольких доз изучаемого лекарства субъектами исследования. Также возможно использовать параллельный дизайн в случае длительного периода полувыведения лекарства и необходимости чрезмерно длительного периода отмывки между периодами исследований с перекрестным дизайном.
Однако исследование с параллельным дизайном потребует значительно большей выборки (см.
81

таблицу 11), и в случае коэффициента вариабельности больше 15% (необходимо включение более 72 субъектов) будет экономически нецелесообразным.
В случае отсутствия данных о коэффициенте внутрииндивидуальной вариабельности конкретного лекарственного средства (в рамках международного непатентованного наименования) возможно рассмотреть вариант исследования с двухэтапным дизайном
(например, адаптивным).
Задача выбора дизайна исследования и его обоснование ложится целиком и полностью на плечи разработчика и/или спонсора.
С регуляторной точки зрения, при любом из перечисленных возможных дизайнов необходим подход, направленный на прямое сужение границ биоэквивалентности (простой перекрестный или параллельный дизайн) или подход с масштабированием границ и сравнительной оценкой внутрииндивидуальной вариабельности воспроизведенного и референтного препаратов в случае повторного дизайна исследования.
При расчете размера выборки необходимо иметь ввиду, что препараты с УТД по умолчанию не могут быть высоковариабельными. Расчет размера выборки в зависимости от дизайна исследования выполняют в специализированных статистических программах согласно следующим рекомендациям: точечная оценка (PE) должна находиться в пределах 95,00% –
105,00% (0,95-1,05); оценка по максимальному значению CVintra для Cmax или AUC; мощность исследования (p) должна быть не менее 80%, ошибка второго рода (β) соответственно 20% (β =1- p), ошибка первого рода (α) должна быть 5%.
В таблицах 13, 14, 15 представлены результаты оценки необходимого объема выборки для исследований с простым перекрестным дизайном, с параллельным дизайном и полностью повторным дизайном [78].
Таблица 13. Размер выборки в зависимости от коэффициента внутрииндивидуальной вариабельности (CVintra) и предполагаемой точечной оценки (PE) при значении ошибки первого рода α=0,05, мощности 1–β=0,8 и допустимых границах доверительного интервала
90,00%–111,11%, стандартный дизайн.
|
|
µT/µR (PE) |
|
||
|
|
|
|
|
|
CVintra |
0,95 |
|
1 |
|
1,05 |
|
|
|
|
|
|
5 |
14 |
|
6 |
|
12 |
|
|
|
|
|
|
7,5 |
26 |
|
12 |
|
24 |
|
|
|
|
|
|
10 |
44 |
|
18 |
|
42 |
|
|
|
|
|
|
12,5 |
68 |
|
26 |
|
64 |
|
|
|
|
|
|
15 |
96 |
|
38 |
|
92 |
|
|
|
|
|
|
17,5 |
130 |
|
50 |
|
124 |
|
|
|
|
|
|
20 |
168 |
|
64 |
|
160 |
|
|
|
|
|
|
22,5 |
212 |
|
80 |
|
200 |
|
|
|
|
|
|
25 |
258 |
|
96 |
|
246 |
|
|
|
|
|
|
|
82 |
|
|
|
|
|
|
µT/µR (PE) |
|
||
|
|
|
|
|
|
CVintra |
0,95 |
|
1 |
|
1,05 |
|
|
|
|
|
|
27,5 |
310 |
|
116 |
|
294 |
|
|
|
|
|
|
30 |
366 |
|
136 |
|
348 |
|
|
|
|
|
|
Таблица 14. Размер выборки в зависимости от коэффициента внутрииндивидуальной вариабельности (CVintra) и предполагаемой точечной оценки (PE) при значении ошибки первого рода α=0,05, мощности 1–β=0,8 и допустимых границах доверительного интервала
90,00%–111,11%, параллельный дизайн.
|
|
µT/µR (PE) |
|
||
|
|
|
|
|
|
CVintra |
0,95 |
|
1 |
|
1,05 |
|
|
|
|
|
|
5 |
24 |
|
6 |
|
12 |
|
|
|
|
|
|
7,5 |
50 |
|
20 |
|
48 |
|
|
|
|
|
|
10 |
86 |
|
34 |
|
82 |
|
|
|
|
|
|
12,5 |
134 |
|
50 |
|
126 |
|
|
|
|
|
|
15 |
190 |
|
72 |
|
180 |
|
|
|
|
|
|
17,5 |
258 |
|
96 |
|
244 |
|
|
|
|
|
|
20 |
334 |
|
124 |
|
316 |
|
|
|
|
|
|
22,5 |
420 |
|
156 |
|
398 |
|
|
|
|
|
|
25 |
516 |
|
192 |
|
488 |
|
|
|
|
|
|
27,5 |
620 |
|
230 |
|
586 |
|
|
|
|
|
|
30 |
732 |
|
270 |
|
692 |
|
|
|
|
|
|
Таблица 15. Размер выборки в зависимости от коэффициента внутрииндивидуальной вариабельности (CVintra) и предполагаемой точечной оценки (PE) при значении ошибки первого рода α=0,05, мощности 1–β=0,8 и границах доверительного интервала 80,00%– 125,00%, полный повторный дизайн (расчеты приведены при предположении, что
вариабельность исследуемого и референтного препарата будут одинаковы).
|
|
µT/µR (PE) |
|
|
|
|
|
CVintra |
0,95 |
1 |
1,05 |
|
|
|
|
7,5 |
70 |
14 |
58 |
|
|
|
|
10 |
34 |
14 |
30 |
|
|
|
|
12,5 |
24 |
14 |
22 |
|
|
|
|
15 |
20 |
14 |
20 |
|
|
|
|
17,5 |
18 |
14 |
18 |
|
|
|
|
20 |
18 |
14 |
16 |
|
|
|
|
22,5 |
18 |
14 |
18 |
|
|
|
|
25 |
18 |
16 |
18 |
|
|
|
|
27,5 |
20 |
16 |
20 |
|
|
|
|
30 |
22 |
18 |
20 |
|
|
|
|
Примечание: расчет не приводится для вариабельности менее 7,5, т.к. данный метод расчета имеет ограничение в случае крайне низкой вариабельности, т.к. при ожидаемом отношении между исследуемым и референтным препаратом в 5% (РЕ 0,95-1,05) размер требуемой выборки нерационально большой.
Таким образом, для препаратов с УТД с целью соответствия данных ЛП критериям
83

признания БЭ 90,00-111,11% в исследованиях со стандартным или параллельным дизайном требуется участие большего числа субъектов чем для большинства препаратов, не относящихся к препаратам с УТД или ВВП. Меньшее количество добровольцев может оказаться достаточным для получения статистически значимых результатов и подтверждения БЭ при использовании повторного (репликативного) дизайна исследования, который, как и в случае препаратов с высокой вариабельностью позволяет значительно уменьшить количество добровольцев (см.
таблицу 15). Приведенный в работе метод расчета размера выборки (программа R, пакет «Power TOST», функция «sampleN.NTIDFDA»[357]) ограничен вариабельностью и ожидаемой точечной оценкой (разностью между исследуемым и референтным препаратом) - 5%, при значениях вариабельности менее 5% или увеличении РЕ более 5% размер выборки нерационально большой
[78].
3.1.2.3 Выбор границ признания биоэквивалентности
Висследованиях БЭ препаратов с УТД со стандартным или параллельным дизайном - отношение ФК параметров AUC0–t (AUC0–∞) или AUC0-72 и Сmax исследуемого препарата к референтному должно лежать в интервале 90,00–111,11 % при 90 %-ном доверительном интервале.
Вслучае использования подхода, предложенного FDA - для препаратов с УТД допускается масштабирование границ для ФК параметров в зависимости от значения σWR (оценка внутрииндивидуальной вариабельности CVintra) референтного препарата, полученного в исследовании с повторным дизайном (см. разделы 3.1.2.4 - 3.1.2.5).
Взависимости от значения σWR масштабирование границ 90 % доверительного интервала для ФК параметров возможно только в пределах 80,00 – 125,00, что равно CVintra≥30%. Если CVintra окажется менее 10% (σWR = 0,0997) - границы признания БЭ при масштабировании окажутся менее 90,00-111,11%, если CVintra будет в диапазоне 10-30% (σWR: 0,0997 – 0,293) - границы масштабируются и являются шире, чем 90,00-111,11%, но уже чем 80,00-125,00% [78].
3.1.2.4 Статистический анализ
Статистическая гипотеза
Модель ABE
В отношении препаратов c УТД в случае исследований с простым перекрестным или параллельным дизайном, применяются нулевая (μT/μR < θL или μT/μR > θU) и альтернативная (θL ≤
μT/μR ≤ θU) гипотезы. Где μT/μR — это отношение генеральных средних исследуемого и референтного препаратов в исходных единицах измерения; θL и θU – нижняя и верхняя принятые допустимые границы БЭ. Нулевая гипотеза заключается в неэквивалентности сравниваемых препаратов, т.е. в доказательстве того, что различия существуют. Альтернативная гипотеза заключается в эквивалентности препаратов, т.е. что различия между препаратами отсутствуют.
84

В соответствии с этим задача исследования БЭ отклонить нулевую гипотезу в пользу альтернативной [78].
В случае проведения логарифмического преобразования формулировка гипотез также трансформируется. Отношение μT/μR преобразуется в разность lnμT-lnμR, при этом по результатам исследования требуется показать, что
- θlnL ≤ μlnT - μlnR ≤ θlnU.
Это уравнение эквивалентно уравнению:
(μlnT – μlnR)2 ≤lnθ2А, т.е. θА = θU = е-lnθL.
Применительно к препаратам с УТД, по-другому ее можно представить следующим
образом:
-ln(0,9) ≤ (μlnT – μlnR) ≤ ln(1,1111)
или
-0,10 ≤ (μlnT – μlnR) ≤ 0,10
или
(μlnT – μlnR)2 ≤ 0,102
̅̅̅ |
̅̅̅ |
|
|
|
√ ( |
1 |
|
1 |
̅̅̅ |
̅̅̅ |
|
|
|
√ ( |
1 |
1 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||
|
|
̂ |
|
+ |
|
|
|
̂ |
|
+ )] |
||||||||
[ − − |
−2 |
|
|
) ; − + |
+ 2−2 |
|
||||||||||||
|
|
1− , 1+ 2 |
|
1 |
|
2 |
|
|
1− , 1 |
|
1 |
|
2 |
|||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||
А=1 при параллельном дизайне и 0,5 при перекрестном. t1-α,n1+n2-2 – значение квантиля распределения Стьюдента
Модель RSABE
Модель применима только для исследований с повторным (репликативным) дизайном.
Как и в отношении высоковариабельных ЛП уравнение θlnL ≤ μlnT - μlnR ≤ θlnU, с учетом повторного дизайна и возможностью определить внутрииндивидуальную вариабельность референтного препарата, приобретает следующий вид:
- θs ≤ (μlnT – μlnR)/σWR ≤ θs,
где σWR — оценка внутрииндивидуальной вариабельности фармакокинетических параметров (AUC0–t (AUC0–∞) или AUC0-72 и Сmax) референтного препарата.
или
- θsσWR ≤ μlnT – μlnR ≤ θsσWR.
или
(μlnT – μlnR)2 ≤ θ2sσWR 2
Сучетом того, что границы для препаратов с УТД должны быть сужены до 90,00-111,11%,
иσW0 не должна превышать 0,1 (регуляторные ограничения для препаратов с УТД):
85

= ,
0
где
= е− ; = 1⁄ ; = 1⁄0,9
Подставив соответствующе значение θA в предыдущее уравнение, мы получим:
(μlnT – μlnR)2 ≤ (lnθ2A/σ2W0)σ2WR.
Правую часть уравнения можно перенести в левую и в итоге альтернативная гипотеза приобретает следующий вид
(μlnT – μlnR)2 - (lnθ2A/σ2W0)σ2WR ≤ 0
Данная гипотеза тестируется при α = 0,05, нам необходимо получить 95% односторонний доверительный интервал (1-α) для верхней границы БЭ (μlnT – μlnR)2 - (lnθ2A/σ2W0)σ2WR. Для расчета верхней границы рекомендуется использовать метод аппроксимации по Хоу (Howe’s Approximation I).
После того как мы получим значение верхней границы 95% доверительного интервала θU,
мы можем рассчитать нижнюю границу θL.
= −
Помимо этого, также необходимо провести проверку критериев БЭ и в стандартных границах признания БЭ 80,00-125,00% [78].
3.1.2.5 Оценка результатов
В случае стандартного простого перекрестного дизайна исследования ЛП с УТД должны оцениваться в границах БЭ 90,00-111,11%, и в случае, если границы их оцененных 90%
доверительных интервалов для значений AUC0-t (AUC0–∞) и Cmax находятся в указанных пределах они признаются БЭ.
Также для ЛП с УТД в последние годы разработаны новые критерии к их изучению и экспертизе - БЭ для них должна определяться с использованием подхода с масштабированием границ признания БЭ в четырехпериодном, полностью повторном (полностью
«репликативным»), перекрестном исследовании, что позволяет одновременно сравнивать эквивалентность ФК параметров AUC и Cmax по их средним значениям и внутрииндивидуальную вариабельность этих параметров у исследуемого и референтного препарата [240, 448]. Данный подход дает возможность масштабировать границы признания БЭ. Масштабирование проводиться на основании полученных в исследовании БЭ с полным повторным дизайном данных о внутрииндивидуальной вариабельности референтного ЛП (σWR). Базовые границы признания БЭ для лекарств с УТД устанавливаются на уровне 90,00-111,11% при внутрииндивидуальной вариабельности - 10%, далее эти границы масштабируются.
86

Чем меньше σWR, тем уже границы БЭ для исследуемого лекарства. Чем выше σWR, тем шире границы БЭ для исследуемого лекарства. То есть если значение внутрииндивидуальной вариабельности референтного ЛП меньше или равно 10%, то масштабированные границы уже,
чем 90,00-111,11%. Если это значение больше 10%, то полученные границы больше, чем 90,00111,11%.
При этом обязательна проверка БЭ по AUC и Cmax в стандартных границах 80,00-125,00%
с целью ограничения данного подхода для препаратов с ВВВ (коэффициент внутрииндивидуальной вариабельности (CVintra) превышает 30%), т.к. основное требование к лекарствам с УТД – это их не высокая внутрииндивидуальная вариабельность. Т.к. граница признания БЭ 80,00-125,00% подразумевает возможность наличия 20% различий между исследуемым и референтным ЛП, которые считаются клинически не значимыми. Однако в случае лекарств с УТД, данное допущение неприемлемо [78].
Таким образом, все воспроизведённые ЛП с УТД должны подтвердить БЭ и в стандартных границах признания БЭ, и при их масштабировании [448].
В упрощенном виде данный подход можно представить следующим алгоритмом:
Шаг 1: Расчет внутрииндивидуальной вариабельности референтного препарата -
sWR.
 =
=
Где, m – число последовательностей; - ni – число субъектов в последовательности i;
Dij=Rij1-Rij2 (Rij1-Rij2 – разница между двумя наблюдениями данного субъекта при приеме R
препарата для субъекта j в последовательности i);
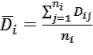 , и
, и 
s2WT - может быть вычислена таким же образом, используя разницу между двумя наблюдениями данного субъекта при приеме Т препарата.
Шаг 2: Масштабирование границ БЭ.
Для исследования БЭ с масштабированием границ признания на основании данных референтного препарата, нулевая и альтернативная гипотезы описываются уравнениями и,
соответственно:
87

Где µТ и µR являются средними логарифмически-трансформированными (натуральными логарифмами) ФК параметров (таких как AUC и Cmax) для исследуемого (T) и референтного (R)
препаратов, соответственно; σWR является внутрииндивидуальным стандартным отклонением для препарата R; и θ является масштабируемой границей признания БЭ в среднем (θ >0).
Отношения между GMR и (μТ - μR) может быть выражено следующим выражением lnGMR=μТ - μR
Альтернативная гипотеза может быть переписана в виде
Кроме того,
где является верхней границей БЭ (должно быть равно 1.11111 (отношение 1/0.9); GMR (отношения геометрических средних исследуемого и рефренного препаратов (T/R)), а σW0 -
является регуляторной константой (в FDA принято значение 0.10).
Проверка этой гипотезы заключается в том, чтобы получить 1-α (т.е., 95%) верхний доверительный предел для величины (μT - μR)2-θ×σ2WR и отклонить H0 в пользу H1, если этот доверительный предел окажется меньше или равен нулю. Способом получения верхнего доверительного предела является аппроксимация I по Хоу (Howe’s approximation I) [240].
Шаг 3: Проверка БЭ в стандартных границах 80,00-125,00%.
Шаг 4: Сравнительная оценка внутрииндивидуальной вариабельности исследуемого
и референтного препарата.
Сравнение σWT/σWR осуществляется с помощью одностороннего F теста. Нулевая гипотеза для этого теста:
H0: σWT/σWR>δ
А альтернативная гипотеза:
H1: σWT/σWR≤δ,
Где σWT является внутрииндивидуальным стандартным отклонением для препарата T и δ является регуляторным пределом для демонстрации, что внутрииндивидуальная вариабельность
T не выше, чем у R. Доверительный интервал (1- α)100% для σWT/σWR задается формулой:
Здесь α=0,1, Fα/2(ν1, ν2) и F1-α/2(ν1, ν2) являются значениями F распределения с ν1
(числитель) и ν2 (знаменатель) степеней свободы, которые имеют вероятность α/2 и 1-α/2 справа
88
от нее, соответственно. sWT – оценка σWT со степенью свободы ν1, sWR – оценка σWR со степенью свободы ν2.
Верхняя граница рассчитанного 90% доверительного интервала для σWT/σWR должна быть меньше или эквивалентна 2.5 [78].
Из результатов проведенного нами анализа можно сделать вывод о том, что наиболее приемлемыми являются подход с сужением границ признания БЭ до 90,00-111,11% при простом перекрестном дизайне исследования, описанный в «Руководстве по экспертизе лекарственных средств», а также подход FDA, предусматривающий использование полного повторного дизайна исследований с масштабированием границ признания БЭ.
Следует отметить, что подход с масштабированием границ, рекомендуемый FDA, является более либеральным и отвечает современным тенденциям в исследованиях БЭ ЛП, в том числе в отношении высоковариабельных препаратов [43, 62].
Данные подходы целесообразно применять для ЛП, перечисленных в таблице 12 и
отнесенных различными регуляторами к препаратам с УТД. Также следует ориентироваться на разработанный нами перечень (см. Приложение №4).
3.1.3. Лекарственные препараты аналоги эндогенных соединений
В отношении ЛП-АЭС в настоящее время в Российской Федерации отсутствуют нормативно-правовые акты, которые регулируют разработку протоколов и правила проведения сравнительных ФК исследований in vivo для таких препаратов. Целью теоретического анализа было осветить проблемы, связанные с изучением и государственной регистрацией ЛП,
являющихся АЭС в Российской Федерации и подготовить рекомендации к планированию и оценке результатов исследований БЭ [79].
3.1.3.1 Определение препаратов аналогов эндогенных соединений
ЛП, действующим веществом которых являются соединения близкие или эквивалентные веществам, синтезирующимся в организме человека вследствие протекающих в нем физиологических и (или) патологических процессов и которые, как правило, являются биологически активными веществами, участвующими в различных метаболических процессах организма, называются препаратами аналогами эндогенных соединений [79].
Для оценки ФК, относительной биодоступности и БЭ ЛП-АЭС, необходимо учитывать природу каждого отдельного эндогенного вещества, механизмы регуляции его гомеостаза, а также то, что в организме указанные соединения всегда присутствуют в фоновых эндогенных концентрациях [79].
Согласно данным отечественной и зарубежной литературы [1, 42, 75, 381] для каждого препарата, содержащего конкретное действующее вещество – аналогичное эндогенному, необходим индивидуальный подход, т.к. нет эталонного решения проблемы фонового
89

содержания эндогенных соединений, но выработаны некоторые ключевые положения, которые следует в обязательном порядке учитывать при проведении исследований БЭ препаратов АЭС
[79].
3.1.3.2 Дизайн исследования и субъекты исследования
В целом для определения оптимального дизайна исследования БЭ каждого препарата-
АЭС следует всесторонне оценивать всю имеющуюся информацию, так как недостаточная оценка может привести к заключению о биоэквивалентности препаратов, когда на самом деле они не биоэквивалентны и наоборот.
В случае отсутствия общедоступных научных данных в отношении того, что прием препарата повышает общее содержание эндогенного вещества, то это следует подтвердить либо в пилотном исследовании, либо в рамках одного из этапов двухэтапного исследования БЭ. При этом следует по возможности изучить различные дозировки референтного препарата, чтобы определить взаимосвязь «доза–концентрация» (т.к. в случае нелинейности ФК лекарственного вещества и высокой эндогенной концентрации может быть ошибочно продемонстрирована БЭ для различных дозировок) [79].
Для препаратов, которые после приема, приводят к незначительному увеличению концентрации по отношению к эндогенной концентрации (например, ≤20%), проведение исследования БЭ не имеет смысла, и в данном случае целесообразно провести сравнительные клинические исследования [16]. Также стоит отметить, что для препаратов, выводящихся из организма главным образом с мочой, почечный клиренс – полезный ФК параметр, отражающий как концентрацию соединения в плазме крови, так и экскрецию. Если почечный клиренс сильно меняется вместе с дозой, то кинетика выведения дозoзависима [175].
Определять эндогенную концентрацию вещества в плазме крови следует на каждом этапе исследования и вычитать ее из общей концентрации измеряемого соединения (проводить коррекцию на эндогенную концентрацию) для каждого субъекта. Эндогенная концентрация определяется всегда до приема исследуемых ЛП [79].
Если соединение синтезируется в организме и по данным литературы известно, что эндогенная концентрация вещества имеет вариабельность, то рекомендуется проводить несколько измерений исходных концентраций эндогенного соединения и учитывать среднюю эндогенную концентрацию.
Для получения максимально точной оценки эндогенного уровня следует определять эндогенные концентрации в тех же точках, в которых будет оцениваться концентрация после приема исследуемых препаратов. То есть схема отбора крови для оценки эндогенного уровня должны полностью совпадать со схемой отбора крови после приема ЛП. Дни определения эндогенной концентрации и дни определения концентраций лекарственного препарата не
90