

4.5 Взаимодействие растворенного в металле кислорода с введенным в него раскислителем
Большинство исследователей считают, что при обычных для производства стали температурах сам акт химического взаимодействия растворенных в металле кислорода и раскислителя, определяемый его энергией активации и кон- центрациями (или активностями) реагирующих веществ, совершается слишком быстро для того, чтобы он мог быть "ведущим" или "тормозящим" звеном процесса раскисления. Однако процессы раскисления металла протекают не только в момент ввода раскислителей при соответствующей температуре, но и при охлаждении еще жидкого металла, при его кристаллизации и, в какой-то мере, при дальнейшем охлаждении уже затвердевшего металла. В этих случаях кинетика процесса, "ведущий" его этап могут меняться.
Количество оксидных включений, которые могут образоваться, еще при охлаждении жидкого металла можно оценить исходя из того, что потенциальная возможность прохождения химического акта взаимодействия кислорода и раскислителя определяется разностью:
![]() , (116)
, (116)
где индексы "наб" относятся к наблюдаемым значениям концентраций или активностей раскислителя и кислорода, а индексы "р" -соответственно, к равновесным. Например, по таблице для раскисления алюминием [123]:
при t=1600 °С kAl=0,4610-14;
при t=1500 °C kAl=0,5710-16,
что и определяет дополнительное количество Аl2O3, образующейся при этом охлаждении. Аналогично для кремния:
при t=1600 °С kSi=0,22710-5;
при t=1600 С kSi=0,27410-6.
Таким образом, чем ниже концентрации 0наб после ввода раскислителей, т.е. чем сильнее (или "глубже") раскисление, тем меньше величина ОR и тем меньшее значение имеет образование так называемых "вторичных" эндогенных продуктов раскисления [22].
4.6 Образование зародышей новой фазы продуктов раскисления в объеме жидкого металла
По мнению большинства исследователей, образование новой фазы продуктов раслисления, в отличие от двух первых этапов этого процесса, часто является ведущим звеном, определяющим скорость раскисления. Эта точка зрения основана на работах Фольмера и Томсона [2,31,124], которые считали, что элементарный процесс
m[R]+n[R](RmOn)
проходит в направлении верхней стрелки и становится необратимым лишь в том случае, когда количество частиц RmOn, одновременно образующихся в одном и том же месте достаточно велико и достигает некоторого «критического» значения. Такой критический зародыш термодинамически достаточно устойчив и уже образовавшись он может расти дальше за счет прогрессирующего формирования RmOn на его поверхности или вблизи ее. При этом необходима затрата энергии на формирование межфазной границы, образуемое включение – маточный расплав (жидкий металл).
Математические зависимости, определяющие работу W и изобаро-изотермический потенциал G образования зародышей новой фазы продуктов раскисления, а также интенсивность их образования имеют вид [45, 138, 139]:
![]() G*=
G*=![]() ; (117)
; (117)
j=Z
exp![]() , (118)
, (118)
где z – частотный фактор, определяющий частоту протекания элементарных процессов;
м-в – межфазное натяжение на границе металла и возникающего в нем зародыша;
М – молекулярный вес образующейся новой фазы;
а – величина пересыщения металла кислородом и раскислителем, т.e.
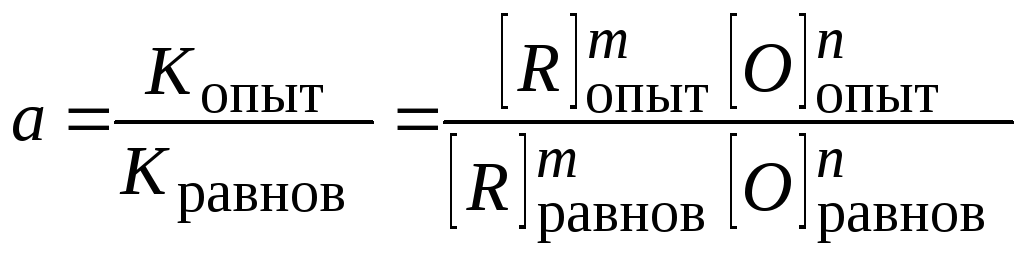 . (119)
. (119)
Величина Z определяется уравнением:
![]() , (120)
, (120)
где k – константа Больцмана; (k=1,3810–23 Дж/К);
h – константа Планка; (h=6,6310–34 Дж/C);
V – объем, приходящийся на 1 атом образующейся фазы;
n – число атомов, присутствующих на поверхности критического зародыша;
n – число атомов, приходящихся на единицу объема исходной фазы.
Величину "J" определяет главным образом экспонента (G/kT), т.е. в конечном счете величина межфазного натяжения м-в, которая по имеющимся экспериментальным данным составляет [136]:
для FеО и сплавов FеO+MnO: 200300 (мДж/м2);
для кремнезема (кварца): 10001300 (мДж/м2);
для силикатов железа и марганца: 500600 (мДж/м2) ;
для герценита: I600I800 (мДж/м2);
для глинозема: 20002100(мДж/м2).
При таких больших значениях м-в интенсивность зарождения включений может быть обеспечена только за счет весьма значительных величин пересыщения металла кислородом и раскислителем "а". Например, по расчетам [138] при значениях м-в равных 100 мДж/м2 и 500 мДж/м2 (т.е. при сравнительно низких) нужны пересыщения "а" порядка 2-3 для вероятной (практически наблюдаемым количествам микроскопических включений) интенсивности их зарождения. По другим подсчетам [139], выполненным для м-в=700 и 100 мДж/м2 величины "J" при данных значениях "а" неизмеримо меньше.
Эти выводы качественно подтверждены рядом исследователей [135,134], хотя проконтролировать их экспериментально по количеству неметаллических включений в опытных образцах или даже на холодных моделях очень сложно. Даже в тщательно поставленных экспериментах неизбежны следующие источники ошибок:
-
Зародыши образующейся новой фазы, состоящие из нескольких сотен или тысяч атомов, очевидно, по физическим свойствам не полностью адекватны с макрообъемами этой фазы, а значения м-в экспериментально найдены именно для макрообьемов этих фаз. Во всяком случае тождественность этих свойств пока не доказана.
-
Микролокальная неравномерность состава даже очень хорошо перемешиваемого металла, которая несомненно влияет на ход реакции и кинетику зарождения ее продуктов.
-
Изменение химического состава микрообьемов металла, окружающих формирующиеся зародыши, вследствие перехода в них растворенных в металле и участвующих в реакции компонентов, приводящее к изменению величины м-в. Это имеет особое значение при низких концентрациях (или активностях.) растворенного кислорода, т.е. при использовании сильных раскислителей.
-
Oдновременно со спонтанным зарождением "гомогенных" включений происходит также энергетически более легкое отложение их на "подложках", т.е. практически неустранима возможность "гетерогенного" формирования включений.
Экспериментальная проверка гипотезы Фольмера-Томсона и основанных на ней расчетов также сопряжена с большими практическими трудностями:
-
Невозможность выполнения микролокальных анализов металла, даже при использовании микрозондов для определения активности растворенного кислорода;
-
Чрезвычайная трудность фиксации состояния металла в момент зарождения в нем неметаллических включений и устранения зарождения в нем неметаллических включений и устранения последующих "вторичных" явлений, происходящих в жидком расплаве при последующих его охлаждении и кристаллизации;
-
Учет количества образующихся зародышей чрезвычайно сложен и недостаточно надежен даже при использовании таких современных инструментов как растровая электронная микроскопия и специализированные счетные устройства[141].
Из сказанного выше и на основании большого количества опытных данных следует, что общая теория спонтанного гомогенного зарождения в стали неметаллических включений и основанные на ней представления о зависимости интенсивности зарождения включений от м-в, а=kопыт/kравен, и т.п. в основном правильны. Однако их количественные оценки и основанные на них расчеты сейчас нельзя еще считать надежными и не следует преувеличивать их значение.
В качестве примера ниже представлены некоторые данные из ряда работ [136, 142, 151], посвященных в основном гомогенному зарождению силикатных неметаллических включений, которое имеет место при затвердевании сплавов Fe-O-Si, т.е. при условиях осложненных ликвационными явлениями, возникновением новых поверхностей кристалл-расплав и изменением температуры. Применив ряд новых и уточненных методов исследовании, авторы [142] пришли к заключению:
-
гомогенное зарождение оксидной фазы в системе Fe-O-Si в определенных условиях, возможно;
-
для этого необходим кислородный потенциал намного больший, чем требуется по условиям равновесия;
-
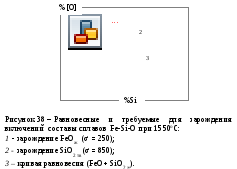
для зарождения в расплаве Fe-O-Si неметаллических силикатных включений необходимо пересыщение, которое при температуре 1550 С составляет:
 80.
80.
Оно остается практически постоянным при изменениях [Si]рав или [O] (см. рис. 38)
-
в расплавах, содержащих менее 0,02 % кислорода, гомогенное зарождение силикатных включений не происходит;
-
в благоприятных условиях в течение первой секунды после достижения равномерного распределения компонентов в расплаве Fe-Si-O образуется огромное количество мельчайших зародышей неметаллических включений (z108 включ/см3).