

2. Специфика белка фибрилл амилоида позволяет выделить al-, аа-, af- и asc1-
амилоидоз.
AL-амилоидоз включает первичный (идиопатический) амилоидоз и амилоидоз при
«плазмоклеточной дискразии», которая объединяет парапротеинемические лейкозы
(миеломная болезнь, болезнь Вальденстрема, болезнь тяжелых цепей Франклина),
злокачественные лимфомы и др. AL-амилоидоз всегда генерализованный с поражением
сердца, легких и сосудов. АА-амилоидоз охватывает вторичный амилоидоз и две формы
наследственного - периодическую болезнь и болезнь Маккла и Уэллса. Это также
генерализованный амилоидоз, но с преимущественным поражением почек. AF-амилоидоз -
наследственный, представлен семейной амилоидной нейропатией (FAP); поражаются прежде
всего периферические нервы. ASC-амилоидоз - старческий генерализованный или системный
(SSA) с преимущественным поражением сердца и сосудов.
3. Учитывая распространенность амилоидоза, различают генерализованную и локальную
формы. Кгенерализованному амилоидозу, как это видно уже из сказанного, относят
первичный амилоидоз и амилоидоз при «плазмоклеточной дискразии» (формы AL-
амилоидоза), вторичный амилоидоз и некоторые типы наследственного (формы АА-
амилоидоза), а также старческий системный амилоидоз (ASC^-амилоидоз).Локальный
амилоидоз
79
объединяет ряд форм наследственного и старческого амилоидоза, а также локальный
опухолевидный амилоидоз («амилоидная опухоль»).
4. Своеобразие клинических проявлений в связи с преимущественным поражением
органов и систем позволит выделять кардиопатический, нефропатический,
нейропатический, гепатопатический, эпинефропатический, смешанный типы амилоидоза
и APUD-амилоидоз. Кардиопатический тип, как говорилось ранее, чаще встречается при
первичном и старческом системном амилоидозе, нефропатический - при вторичном
амилоидозе, периодической болезни и болезни Маккла и Уэллса; для вторичного амилоидоза
характерны и смешанные типы (сочетание поражения почек, печени, надпочечников,
желудочно-кишечного тракта). Нейропатический амилоидоз, как правило, имеет
наследственный характер. APUD-амилоид развивается в органах APUD-системы при
развитии в них опухолей (апудом), а также в островках поджелудочной железы при
старческом амилоидозе.
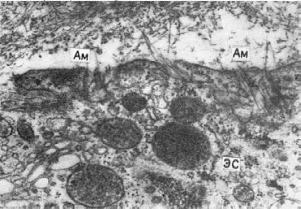
Морфо- и патогенез амилоидоза. Функцию амилоидобластов, продуцирующих белок
фибрилл амилоида (рис. 34), при различных формах амилоидоза выполняют разные клетки.
При генерализованных формах амилоидоза - это главным образом макрофаги,
плазматические и миеломные клетки; однако не исключается роль фибробластов,
ретикулярных клеток и эндотелиоцитов. При локальных формах в роли амилоидобластов
могут выступать кардиомиоциты (амилоидоз сердца), гладкие мышечные клетки (амилоидоз
аорты), кератиноциты (амилоидоз кожи), В-клетки островков поджелудочной железы
(инсулярный амилоидоз), С-клетки щитовидной железы и другие эпителиальные клетки
APUD-системы.
Рис.
34. Амилоидобласт. Фибриллы амилоида (Ам) в инвагинатах плазмолеммы звездчатого
ретикулоэндотелиоцита с гиперплазией гранулярной эндоплазматической сети (ЭС),
свидетельствующей о его высокой синтетической активности. х30 000
80
Появление клона амилоидобластов объясняет мутационная теория амилоидоза (Серов
В.В., Шамов И.А., 1977). При вторичном амилоидозе (исключая амилоидоз при
«плазмоклеточной дискразии») мутации и появление амилоидобластов можно связать с
длительной антигенной стимуляцией. Клеточные мутации при «плазмоклеточной дискразии»
и амилоидозе опухолей, а возможно, и при опухолевидном локальном амилоидозе
обусловлены опухолевыми мутагенами. При генетическом (семейном) амилоидозе речь идет
о мутации гена, которая может произойти в различных локусах, чем и определяются различия
в составе амилоидного белка у разных людей и животных. При старческом амилоидозе,
вероятнее всего, имеют место подобные механизмы, так как эту разновидность амилоидоза
рассматривают как фенокопию генетического. Поскольку антигены белка амилоидных
фибрилл являются чрезвычайно слабыми иммуногенами, мутирующиеся клетки не
распознаются иммунокомпетентной системой и не элиминируются. Развивается
иммунологическая толерантность к белкам амилоида, что обусловливает прогрессирование
амилоидоза, чрезвычайно редкое рассасывание амилоида - амилоидоклазия - с помощью
макрофагов (гигантские клетки инородных тел).
Образование амилоидного белка может быть связано с ретикулярными (периретикулярныи
амилоидоз) или коллагеновыми (периколлагеновыи амилоидоз) волокнами.
Для периретикулярного амилоидоза, при котором амилоид выпадает по ходу мембран
сосудов и желез, а также ретикулярной стромы паренхиматозных органов характерно
преимущественное поражение селезенки, печени, почек, надпочечников, кишечника, интимы
сосудов мелкого и среднего калибра (паренхиматозный амилоидоз). Для периколлагенового
амилоидоза, при котором амилоид выпадает по ходу коллагеновых волокон, свойственно
преимущественное поражение адвентиции сосудов среднего и крупного калибра, миокарда,
поперечнополосатой и гладкой мускулатуры, нервов, кожи (мезенхимальный амилоидоз).
Таким образом, амилоидные отложения имеют довольно типичную локализацию: в стенках
кровеносных и лимфатических капилляров и сосудов в интиме или адвентиции; в строме
органов по ходу ретикулярных и коллагеновых волокон; в собственной оболочке железистых
структур. Амилоидные массы вытесняют и замещают паренхиматозные элементы органов,
что ведет к развитию их хронической функциональной недостаточности.
Патогенез амилоидоза сложен и неоднозначен у различных его форм и типов. Лучше других
форм изучен патогенез АА- и AL-амилоидоза.
При АА-амилоидозе фибриллы амилоида образуются из поступающего в макрофаг -
амилоидобласт плазменного предшественника фибриллярного белка амилоида - белка SAA,
который усиленно синтезируется в печени (схема III). Усиленный синтез SAA гепатоцитами
стимулирует макрофагальный медиаторинтерлейкин-1, что приводит к резкому увеличению
содержания SAA в крови (предамилоидная стадия). В этих условиях макрофаги не в
состоянии осуществить полную деградацию SAA, и из
Схема III. Патогенез AA-амилоидоза
81
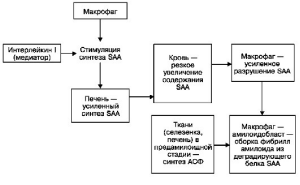
его
фрагментов в инвагинатах плазматической мембраны амилоидобласта происходит сборка
фибрилл амилоида (см. рис. 34). Стимулирует эту сборку амилоидстимулирующий
фактор (АСФ), который обнаруживается в тканях (селезенка, печень) в предамилоидной
стадии. Таким образом, ведущую роль в патогенезе АА-амилоидоза играет макрофагальная
система: она стимулирует усиленный синтез белка предшественника - SAA печенью, она же
участвует и в образовании фибрилл амилоида из деградирующих фрагментов этого белка.
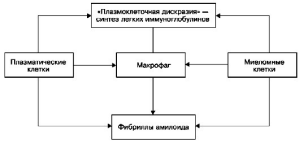
При AL-амилоидозе сывороточным предшественником белка амилоидных фибрилл являются
L-цепи иммуноглобулинов. Считают, что возможны два механизма образования AL-
амилоидных фибрилл: 1) нарушение деградации моноклоновых легких цепей с образованием
фрагментов, способных к агрегации в амилоидные фибриллы; 2) появление L-цепей с
особыми вторичными и третичными структурами при аминокислотных заменах. Синтез
амилоидных фибрилл из L-цепей иммуноглобулинов может происходить не только в
макрофагах, но и в плазматических и миеломных клетках, синтезирующих парапротеины
(схема IV). Таким образом, к патогенезу AL-амилоидоза причастна прежде всего лимфоидная
система; с ее извращенной функцией связано появление «амилоидогенных» легких цепей
иммуноглобулинов - предшественника амилоидных фибрилл. Роль макрофагальной системы
при этом вторичная, соподчиненная.
Макро- и микроскопическая характеристика амилоидоза. Внешний вид органов при
амилоидозе зависит от степени процесса. Если отложения амилоида небольшие, внешний
вид органа изменяется мало и амилоидоз
Схема IV. Патогенез AL-амилоидоза
82
обнаруживается лишь при микроскопическом исследовании. При выраженном амилоидозе
орган увеличивается в объеме, становится очень плотным и ломким, а на разрезе имеет
своеобразный восковидный, или сальный, вид.
В селезенке амилоид откладывается в лимфатических фолликулах (рис. 35) или же
равномерно по всей пульпе. В первом случае амилоидноизмененные фолликулы
увеличенной и плотной селезенки на разрезе имеют вид полупрозрачных зерен,
напоминающих зерна саго (саговая селезенка). Во втором случае селезенка увеличена,
плотная, коричнево-красная, гладкая, имеет сальный блеск на разрезе (сальная
селезенка).Саговая и сальная селезенка представляют последовательные стадии процесса.
В почках амилоид откладывается в стенке сосудов, в капиллярных петлях и мезангии
клубочков, в базальных мембранах канальцев и в строме. Почки становятся плотными,
большими и «сальными». По мере нарастания процесса клубочки и пирамиды полностью
замещаются амилоидом (см. рис. 35), разрастается соединительная ткань и развивается
амилоидное сморщивание почек.
В печени отложение амилоида наблюдается между звездчатыми ретикулоэндотелиоцитами
синусоидов, по ходу ретикулярной стромы долек, в стенках сосудов, протоков и в
соединительной ткани портальных трактов. По мере накопления амилоида печеночные клетки
атрофируются и погибают. При этом печень увеличена, плотная, выглядит «сальной».
В кишечнике амилоид выпадает по ходу ретикулярной стромы слизистой оболочки, а также в
стенках сосудов как слизистой оболочки, так и подслизистого слоя. При резко выраженном
амилоидозе железистый аппарат кишечника атрофируется.
Амилоидоз надпочечников, как правило, двусторонний, отложение амилоида встречается в
корковом веществе по ходу сосудов и капилляров.
83
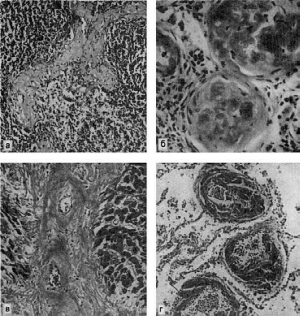
Рис.
35. Амилоидоз:
а - амилоид в фолликулах селезенки (саговая селезенка); б - амилоид в сосудистых клубочках
почек; в - амилоид между мышечными волокнами сердца; г - амилоид в стенках сосудов
легких
В сердце амилоид обнаруживается под эндокардом, в строме и сосудах миокарда (см. рис.
35), а также в эпикарде по ходу вен. Отложение амилоида в сердце ведет к резкому его
увеличению (амилоидная кардиомегалия). Оно становится очень плотным, миокард
приобретает сальный вид.
В скелетных мышцах, как и в миокарде, амилоид выпадает по ходу межмышечной
соединительной ткани, в стенках сосудов и в нервах.
Периваскулярно и периневрально нередко образуются массивные отложения амилоидного
вещества. Мышцы становятся плотными, полупрозрачными.
84
В легких отложения амилоида появляются сначала в стенках разветвлений легочных
артерии и вены (см. рис. 35), а также в перибронхиальной соединительной ткани. Позже
амилоид появляется в межальвеолярных перегородках.
В головном мозге при старческом амилоидозе амилоид находят в сенильных бляшках коры,
сосудах и оболочках.
Амилоидоз кожи характеризуется диффузным отложением амилоида в сосочках кожи и ее
ретикулярном слое, в стенках сосудов и по периферии сальных и потовых желез, что
сопровождается деструкцией эластических волокон и резкой атрофией эпидермиса.
Амилоидоз поджелудочной железы имеет некоторое своеобразие. Помимо артерий железы,
встречается и амилоидоз островков, что наблюдается в глубокой старости.
Амилоидоз щитовидной железы также своеобразен. Отложения амилоида в строме и
сосудах железы могут быть проявлением не только генерализованного амилоидоза, но и
медуллярного рака железы (медуллярный рак щитовидной железы с амилоидозом стромы).
Амилоидоз стромы часто встречается в опухолях эндокринных органов и APUD-системы
(медуллярный рак щитовидной железы, инсулома, карциноид, феохромоцитома, опухоли
каротидных телец, хромофобная аденома гипофиза, гипернефроидный рак), причем в
образовании APUD-амилоида доказано участие эпителиальных опухолевых клеток.
Исход. Неблагоприятный. Амилоидоклазия - исключительно редкое явление при локальных
формах амилоидоза.
Функциональное значение определяется степенью развития амилоидоза. Выраженный
амилоидоз ведет к атрофии паренхимы и склерозу органов, к их функциональной
недостаточности. При выраженном амилоидозе возможна хроническая почечная, печеночная,
сердечная, легочная, надпочечниковая, кишечная (синдром нарушенного всасывания)
недостаточность.
Стромально-сосудистые жировые дистрофии (липидозы)
Стромально-сосудистые жировые дистрофии возникают при нарушениях обмена
нейтральных жиров или холестерина и его эфиров.
Нарушения обмена нейтральных жиров
Нарушения обмена нейтральных жиров проявляются в увеличении их запасов в жировой
ткани, которое может иметь общий или местный характер.
Нейтральные жиры - это лабильные жиры, обеспечивающие энергетические запасы
организма. Они сосредоточены в жировых депо (подкожная клетчатка, брыжейка, сальник,
эпикард, костный мозг). Жировая ткань выполняет не только обменную, но и опорную,
механическую, функцию, поэтому она способна замещать атрофирующиеся ткани.
85
Ожирение, или тучность, - увеличение количества нейтральных жиров в жировых депо,
имеющее общий характер. Оно выражается в обильном отложении жиров в подкожной
клетчатке, сальнике, брыжейке, средостении, эпикарде. Жировая ткань появляется также там,
где она обычно отсутствует или имеется лишь в небольшом количестве, например в строме
миокарда, поджелудочной железе (рис. 36, а). Большое клиническое зна-
Рис.
36. Ожирение:
а - разрастание жировой ткани в строме поджелудочной железы (сахарный диабет); б -
ожирение сердца, под эпикардом толстый слой жира
чение имеет ожирение сердца при тучности. Жировая ткань, разрастаясь под эпикардом,
окутывает сердце, как футляром (рис. 36, б). Она прорастает строму миокарда, особенно в
субэпикардиальных отделах, что ведет к атрофии мышечных клеток. Ожирение обычно резче
выражено в правой половине сердца. Иногда вся толща миокарда правого желудочка
замещается жировой тканью, в связи с чем может произойти разрыв сердца.
Классификация. Она основывается на различных принципах и учитывает причину, внешние
проявления (типы ожирения), степень превышения «идеальной» массы тела,
морфологические изменения жировой ткани (варианты ожирения).
По этиологическому принципу выделяют первичную и вторичную формы ожирения.
Причина первичного ожирения неизвестна, поэтому его называют также
идиопатическим. Вторичное ожирение представлено следующими его видами: 1)
алиментарное, причиной которого является несбалансированное питание и гиподинамия; 2)
церебральное, развивающееся при травме, опухолях мозга, ряде нейротропных инфекций; 3)
эндокринное, представленное рядом синдромов (синдромы Фрелиха и Иценко-Кушинга,
адипозогенитальная дистрофия, гипогонадизм, гипотиреоз); 4) наследственное в виде
синдрома Лоренса-Муна-Бидля и болезни Гирке.
86
По внешним проявлениям различают симметричный (универсальный), верхний, средний и
нижний типы ожирения. При симметричном типе
жиры относительно равномерно откладываются в разных частях тела. Верхний тип
характеризуется накоплением жира преимущественно в области подкожной клетчатки лица,
затылка, шеи, верхнего плечевого пояса, молочных желез. При среднем типе жир
откладывается в подкожной клетчатке живота в виде фартука, при нижнем типе - в области
бедер и голеней.
По превышению массы тела больного выделяют несколько степеней ожирения. При I
степени ожирения избыточная масса тела составляет 20-29%, при II - 30-49%, при III - 50-99%
и при IV - до 100% и более.
При характеристике морфологических изменений жировой ткани при ожирении учитывают
число адипозоцитов и их размер. На этом основании выделяют гипертрофический и
гиперпластический варианты общего ожирения. При гипертрофическом варианте жировые
клетки увеличены и содержат в несколько раз больше триглицеридов, чем обычные; при этом
число адипозоцитов не меняется. Адипозоциты малочувствительны к инсулину, но
высокочувствительны к липолитическим гормонам; течение болезни злокачественное.
При гиперпластическом варианте число адипозоцитов увеличено (известно, что число
жировых клеток достигает максимума в пубертатном периоде и в дальнейшем не меняется).
Однако функция адипозоцитов не нарушена, метаболические изменения их отсутствуют;
течение болезни доброкачественное.
Причины и механизмы развития. Среди причин общего ожирения, как уже говорилось,
большое значение имеют несбалансированное питание и гиподинамия, нарушение нервной
(ЦНС) и эндокринной регуляции жирового обмена, наследственные (семейно-
конституциональные) факторы. Непосредственный механизм ожирения лежит в нарушении
равновесия липогенеза и липолиза в жировой клетке в пользу липогенеза (схема V). Как
видно из схемы V, усиление липогенеза, как и ослабление липолиза,
Схема V. Липогенез и липолиз в жировой клетке
87
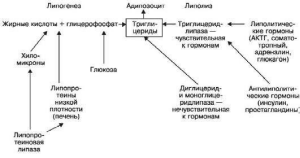
связано не
только с активацией липопротеиновой липазы и угнетением липолитических липаз, но и
нарушением гормональной регуляции в пользу антилиполитических гормонов, состоянием
жирового обмена в кишечнике и печени.
Значение. Будучи проявлением ряда заболеваний, общее ожирение определяет развитие
тяжелых осложнений. Избыточная масса тела, например, является одним из факторов риска
при ишемической болезни сердца.
Исход общего ожирения редко бывает благоприятным.
Антиподом общего ожирения является истощение, в основе которого лежит атрофия.
Истощение наблюдается также в терминальной стадии кахексии (от греч. kakos -
плохой, hexis - состояние).
При увеличении количества жировой клетчатки, имеющем местный характер, говорят
о липоматозах. Среди них наибольший интерес представляет болезнь Деркума (lipomatosis
dolorosa), при которой в подкожной клетчатке конечностей и туловища появляются узловатые
болезненные отложения жира, похожие на липомы. В основе заболевания лежит
полигландулярная эндокринопатия. Местное увеличение количества жировой ткани нередко
является выражением вакатного ожирения (жировое замещение) при атрофии ткани или
органа (например, жировое замещение почки или вилочковой железы при их атрофии).
Антиподом липоматозов служат регионарные липодистрофии, сущность которых состоит в
очаговой деструкции жировой ткани и распаде жиров, нередко с воспалительной реакцией и
образованием липогранулем (например, липогранулематоз при рецидивирующем
ненагнаивающемся панникулите, или болезни Вебера-Крисчена).
Нарушения обмена холестерина и его эфиров
Нарушения обмена холестерина и его эфиров лежат в основе тяжелого заболевания -
атеросклероза. При этом в интиме артерий накапливаются не только холестерин и его
эфиры, но и β-липопротеиды низкой плотности и белки плазмы крови, чему способствует
88
повышение сосудистой проницаемости. Накапливающиеся высокомолекулярные вещества
ведут к деструкции интимы, распадаются и омыляются. В результате этого в интиме
образуется жиробелковый детрит (athere - кашицеобразная масса), разрастается
соединительная ткань (sclerosis - уплотнение) и формируется фиброзная бляшка, нередко
суживающая просвет сосуда (см.Атеросклероз).
Наследственной дистрофией, развивающейся в связи с нарушением обмена холестерина,
является семейный гиперхолестеринемический ксантоматоз. Его относят к болезням
накопления, хотя характер ферментопатии не установлен. Холестерин откладывается в коже,
стенках крупных сосудов (развивается атеросклероз), клапанах сердца и других органах.
Стромально-сосудистые углеводные дистрофии
Стромально-сосудистые углеводные дистрофии могут быть связаны с нарушением баланса
гликопротеидов и гликозаминогликанов. Стромальнососудистую дистрофию, связанную с
нарушением обмена гликопроте-
идов, называют ослизнением тканей. Сущность его состоит в том, что хромотропные
вещества высвобождаются из связей с белками и накапливаются главным образом в
межуточном веществе. В отличие от мукоидного набухания при этом происходит замещение
коллагеновых волокон слизеподобной массой. Собственно соединительная ткань, строма
органов, жировая ткань, хрящ становятся набухшими, полупрозрачными, слизеподобными, а
клетки их - звездчатыми или причудливыми отростчатыми.
Причина. Ослизнение тканей происходит чаще всего вследствие дисфункции эндокринных
желез, истощения (например, слизистый отек, или микседема, при недостаточности
щитовидной железы; ослизнение соединительнотканных образований при кахексии любого
генеза).
Исход. Процесс может быть обратимым, однако прогрессирование его приводит к
колликвации и некрозу ткани с образованием полостей, заполненных слизью.
Функциональное значение определяется тяжестью процесса, его продолжительностью и
характером ткани, подвергшейся дистрофии.
Наследственные нарушения обмена гликозаминогликанов (мукополисахаридов)
представлены большой группой болезней накопления - мукополисахаридозами. Среди них
основное клиническое значение имеетгаргоилизм, или болезнь Пфаундлера-Гурлера, для
которой характерны непропорциональный рост, деформация черепа («массивный череп»),
других костей скелета, наличие пороков сердца, паховой и пупочной грыж, помутнение
роговицы, гепато- и спленомегалии. Считают, что в основе мукополисахаридозов лежит
недостаточность специфического фактора, определяющего обмен гликозаминогликанов.
Смешанные дистрофии
89
О смешанных дистрофиях говорят в тех случаях, когда морфологические проявления
нарушенного метаболизма выявляются как в паренхиме, так и в строме, стенке сосудов
органов и тканей. Они возникают при нарушениях обмена сложных белков -
1
хромопротеидов, нуклеопротеидов и липопротеидов , а такжеминералов.
2
Нарушения обмена хромопротеидов (эндогенные пигментации)
Хромопротеиды - окрашенные белки, или эндогенные пигменты, играют важную роль в
жизни организма. С помощью хромопротеидов осуществляются дыхание (гемоглобин,
цитохромы), выработка секретов (желчь) и инкретов (серотонин), защита организма от
воздействия лучевой энергии (меланин), пополнение запасов железа (ферритин), баланс
витаминов (липохромы) и т.д. Обмен пигментов регулируется вегетативной нервной системой,
эндокринными железами, он тесно связан с функцией органов кроветворения и системы
моноцитарных фагоцитов.
1
Нарушения обмена липопртеидов приведены в разделах о липидогенных пигментах,
жировых и белковых дистрофиях.
2
Помимо эндогенных, существуют экзогенные пигментации (см. Профессиональныне
болезни).
Классификация. Эндогенные пигменты принято делить на 3
группы: гемоглобиногенные, представляющие собой различные производные
гемоглобина, протеиногенные, или тирозиногенные, связанные с обменом тирозина,
и липидогенные, или липопигменты, образующиеся при обмене жиров.
Нарушения обмена гемоглобиногенных пигментов
В норме гемоглобин проходит ряд циклических превращений, обеспечивающих его ресинтез и
образование необходимых для организма продуктов. Эти превращения связаны со старением
и разрушением эритроцитов (гемолиз, эритрофагия), постоянным обновлением эритроцитной
массы. В результате физиологического распада эритроцитов и гемоглобина образуются
пигменты ферритин, гемосидерин и билирубин. В патологических условиях вследствие
многих причин гемолиз может быть резко усилен и осуществляться как в циркулирующей
крови (интраваскулярно), так и в очагах кровоизлияний (экстраваскулярно). В этих условиях,
помимо увеличения образующихся в норме гемоглобиногенных пигментов, может появляться
ряд новых пигментов - гематоидин, гематины и порфирин.
В связи с накоплением гемоглобиногенных пигментов в тканях могут возникать различные
виды эндогенных пигментации, которые становятся проявлением ряда заболеваний и
патологических состояний.
Ферритин - железопротеид, содержащий до 23% железа. Железо ферритина связано с
белком, который носит название апоферритина. В норме ферритин обладает дисульфидной
группой. Это неактивная (окисленная) форма ферритина - SS-ферритин. При недостаточности
кислорода происходит восстановление ферритина в активную форму - SH-ферритин, который
90
обладает вазопаралитическими и гипотензивными свойствами. В зависимости от
происхождения различают анаболический и катаболический ферритин. Анаболический
ферритин образуется из железа, всасывающегося в кишечнике, катаболический - из железа
гемолизированных эритроцитов. Ферритин (апоферритин) обладает антигенными свойствами.
Ферритин образует берлинскую лазурь (железосинеродистое железо) под действием
железосинеродистого калия и соляной, или хлористоводородной, кислоты (реакция Перлса) и
может быть идентифицирован с помощью специфической антисыворотки при
иммунофлюоресцентном исследовании. Большое количество ферритина содержится в
печени (депо ферритина), селезенке, костном мозге и лимфатических узлах, где обмен его
связан с синтезом гемосидерина, гемоглобина и цитохромов.
В условиях патологии количество ферритина может увеличиваться как в тканях, так и в
крови. Повышение содержания ферритина в тканях наблюдается при гемосидерозе, так как
полимеризация ферритина ведет к образованию гемосидерина. Ферритинемией объясняют
необратимость шока, сопровождающегося сосудистым коллапсом, так как SH-ферритин
выступает в роли антагониста адреналина.
Гемосидерин образуется при расщеплении гема и является полимером ферритина. Он
представляет собой коллоидную гидроокись железа, связанную с белками,
гликозаминогликанами и липидами клетки. Клетки, в которых образуется гемосидерин,
называются сидеробластами. В их сидеросомах происходит синтез гранул гемосидерина
(рис. 37). Сидеробласты могут быть как мезенхимальной,
91
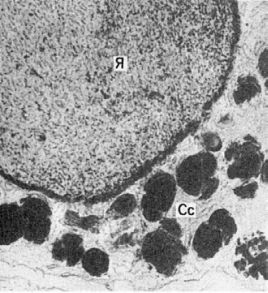
Рис. 37. Сидеробласт.
Крупное ядро (Я), узкий ободок цитоплазмы с большим числом сидеросом (Сс).
Электронограмма. х20 000
так и эпителиальной природы. Гемосидерин постоянно обнаруживается в ретикулярных и
эндотелиальных клетках селезенки, печени, костного мозга, лимфатических узлах. В
межклеточном веществе он подвергается фагоцитозу сидерофагами.
Присутствие в гемосидерине железа позволяет выявлять его с помощью характерных
реакций: образование берлинской лазури (реакция Перлса), турнбулевой сини (обработка
срезов сульфидом аммония, а затем железосинеродистым калием и хлористоводородной
кислотой). Положительные реакции на железо отличают гемосидерин от сходных с ним
пигментов (гемомеланин, липофусцин, меланин).
В условиях патологии наблюдается избыточное образование гемосидерина -
гемосидероз. Он может иметь как общий, так и местный характер.
Общий, или распространенный, гемосидероз наблюдается при внутрисосудистом
разрушении эритроцитов (интраваскулярный гемолиз) и встречается при болезнях системы
кроветворения (анемии, гемобластозы), интоксикациях гемолитическими ядами, некоторых
инфекционных заболеваниях (возвратный тиф, бруцеллез, малярия и др.), переливаниях
иногруппной крови, резус-конфликте и т.д. Разрушенные эритроциты, их обломки, гемоглобин
92
идут на построение гемосидерина. Сидеробластами становятся ретикулярные,
эндотелиальные и гистиоцитарные элементы селезенки, печени, костного мозга,
лимфатических узлов, а также эпителиальные клетки печени, почек, легких, потовых и
слюнных желез. Появляется большое число сидерофагов, которые не успевают поглощать
гемосидерин, загружающий межклеточное вещество. В результате этого коллагеновые и
эластические волокна пропитываются железом. При этом селезенка, печень, костный мозг и
лимфатические узлы становятся ржаво-коричневыми.
Близко к общему гемосидерозу своеобразное заболевание - гемохроматоз, который может
быть первичным (наследственный гемохроматоз) или вторичным.
Первичный гемохроматоз - самостоятельное заболевание из группы болезней накопления.
Передается доминантно-аутосомным путем и связано с наследственным дефектом
ферментов тонкой кишки, что ведет к повышенному всасыванию пищевого железа, которое
в виде гемосидерина откладывается в большом количестве в органах. Обмен железа
эритроцитов при этом не нарушен. Количество железа в организме увеличивается
в десятки раз, достигая 50-60 г. Развивается гемосидероз печени, поджелудочной железы,
эндокринных органов, сердца, слюнных и потовых желез, слизистой оболочки кишечника,
сетчатки глаза и даже синовиальных оболочек; одновременно в органах увеличивается
содержание ферритина. В коже и сетчатке глаз увеличивается содержание меланина, что
связано с поражением эндокринной системы и нарушением регуляции меланинобразования.
Основными симптомами болезни являются бронзовая окраска кожи, сахарный диабет
(бронзовый диабет) и пигментный цирроз печени. Возможно развитие и пигментной
кардиомиопатии с нарастающей сердечной недостаточностью.
Вторичный гемохроматоз - заболевание, развивающееся при приобретенной
недостаточности ферментных систем, обеспечивающих обмен пищевого железа, что ведет
к распространенному гемосидерозу. Причиной этой недостаточности могут быть избыточное
поступление железа с пищей (железосодержащие препараты), резекция желудка,
хронический алкоголизм, повторные переливания крови, гемоглобинопатии
(наследственные,заболевания, в основе которых лежит нарушение синтеза гема или
глобина). При вторичном гемохроматозе содержание железа повышено не только в тканях, но
и в сыворотке крови. Накопление гемосидерина и ферритина, наиболее выраженное в печени,
поджелудочной железе и сердце, приводит кциррозу печени, сахарному
диабету и кардиомиопатии.
Местный гемосидероз - состояние, развивающееся при внесосудистом разрушении
эритроцитов (экстраваскулярный гемолиз), т.е. в очагах кровоизлияний. Оказавшиеся вне
сосудов эритроциты теряют гемоглобин и превращаются в бледные круглые тельца («тени»
эритроцитов), свободный гемоглобин и обломки эритроцитов идут на построение пигмента.
Сидеробластами и сидерофагами становятся лейкоциты, гистиоциты, ретикулярные клетки,
эндотелий, эпителий. Сидерофаги могут долго сохраняться на месте бывшего кровоизлияния,
нередко они переносятся током лимфы в близлежащие лимфатические узлы, где
93
задерживаются, и узлы становятся ржавыми. Часть сидерофагов разрушается, пигмент
высвобождается и в дальнейшем снова подвергается фагоцитозу.
Гемосидерин образуется при всех кровоизлияниях, как мелких, так и крупных. В небольших
кровоизлияниях, которые чаще имеют характер диапедезных, обнаруживается только
гемосидерин. При крупных кровоизлияниях по периферии, среди живой ткани образуется
гемосидерин, а в центре - кровоизлияния, где аутолиз происходит без доступа кислорода и
участия клеток, появляются кристаллы гематоидина.
В зависимости от условий развития местный гемосидероз может возникать в пределах не
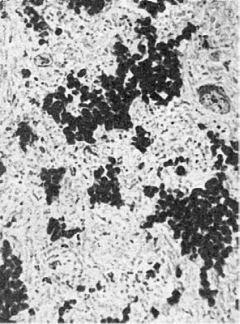
только участка ткани (гематома), но и целого органа. Таков гемосидероз легких,
наблюдающийся при ревматическом митральном пороке сердца, кардиосклерозе и др. (рис.
38). Хронический венозный застой в легких ведет к множественным диапедезным
кровоизлияниям, в связи с чем в межальвеолярных перегородках, альвеолах,
Рис. 38. Гемосидероз легких.
Цитоплазма гистиоцитов и альвеолярного эпителия (сидеробластов и сидерофагов)
нагружена зернами пигмента
94
лимфатических сосудах и узлах легких появляется большое число нагруженных
гемосидерином клеток (см.Венозное полнокровие).
Билирубин - важнейший желчный пигмент. Его образование начинается в гистиоцитарно-
макрофагальной системе при разрушении гемоглобина и отщеплении от него гема. Гем
теряет железо и превращается в биливердин, при восстановлении которого образуется
билирубин в комплексе с белком. Гепатоциты осуществляют захват пигмента, конъюгацию его
с глюкуроновой кислотой и экскрецию в желчные капилляры. С желчью билирубин поступает
в кишечник, где часть его всасывается и вновь попадает в печень, часть - выводится с калом в
виде стер-кобилина и мочой в виде уробилина. В норме билирубин встречается в
растворенном состоянии в желчи и в небольшом количестве в плазме крови.
Билирубин представлен красножелтыми кристаллами. Он не содержит железа. Для
выявления его употребляют реакции, основанные на способности пигмента легко окисляться
с образованием различно окрашенных продуктов. Такова, например, реакция Гмелина, при
которой под воздействием концентрированной азотной кислоты билирубин дает сначала
зеленое, а затем синее или пурпурное окрашивание.
Нарушение обмена билирубина связано с расстройством его образования и выделения. Это
ведет к повышенному содержанию билирубина в плазме крови и желтому окрашиванию им
кожи, склер, слизистых и серозных оболочек и внутренних органов - желтухе.
Механизм развития желтухи различен, что позволяет выделять три ее вида: надпеченочную
(гемолитическую), печеночную (паренхиматозную) и подпеченочную (механическую).
Надпеченочная (гемолитическая) желтуха характеризуется повышенным образованием
билирубина в связи с увеличенным распадом эритроцитов. Печень в этих условиях образует
большее, чем в норме, количество пигмента, однако вследствие недостаточности захвата
билирубина гепатоцитами уровень его в крови остается повышенным. Гемолитическая
желтуха наблюдается при инфекциях (сепсис, малярия, возвратный тиф) и интоксикациях
(гемолитические яды), при изоиммунных (гемолитическая болезнь новорожденных,
переливание несовместимой крови) и аутоиммунных (гемобластозы, системные заболевания
соединительной ткани) конфликтах. Она может развиваться и при массивных кровоизли-
яниях, геморрагических инфарктах в связи с избыточным поступлением билирубина в кровь
из очага распада эритроцитов, где желчный пигмент выявляется в виде кристаллов. С
образованием в гематомах билирубина связано изменение их окраски.
Гемолитическая желтуха может быть обусловлена дефектом эритроцитов. Таковы
наследственные ферментопатии (микросфероцитоз, овалоцитоз), гемоглобинопатии, или
гемоглобинозы (талассемия, или гемоглобиноз F; серповидноклеточная анемия, или
гемоглобиноз S), пароксизмальная ночная гемоглобинурия, так называемые шунтовые
желтухи (при дефиците витамина В12, некоторых гипопластических анемиях и др.).
95
Печеночная (паренхиматозная) желтуха возникает при поражении гепатоцитов, в результате
чего нарушаются захват ими билирубина, конъюгация его с глюкуроновой кислотой и
экскреция. Такая желтуха наблюдается при остром и хроническом гепатитах, циррозах
печени, медикаментозных ее повреждениях и аутоинтоксикации, например при беременности,
ведущих к внутрипеченочному холестазу. Особую группу составляютферментопатические
печеночные желтухи, возникающие при наследственных пигментных гепатозах, при которых
нарушена одна из фаз внутрипеченочного обмена билирубина.
Подпеченочная (механическая) желтуха связана с нарушением проходимости желчных
протоков, что затрудняет экскрецию и определяет регургитацию желчи. Эта желтуха
развивается при наличии препятствий оттоку желчи из печени, лежащих внутри или вне
желчных протоков, что наблюдается при желчнокаменной болезни, раке желчных путей,
головки поджелудочной железы и сосочка двенадцатиперстной кишки, атрезии (гипоплазии)
желчных путей, метастазах рака в перипортальные лимфатические узлы и печень. При застое
желчи в печени возникают очаги некроза с последующим замещением их соединительной
тканью и развитием цирроза (вторичный билиарный цирроз). Застой желчи приводит к
расширению желчных протоков и разрыву желчных капилляров.
Развивается холемия, которая вызывает не только интенсивную окраску кожи, но и явления
общей интоксикации, главным образом от воздействия на организм циркулирующих в крови
желчных кислот (холалемия). В связи с интоксикацией понижается способность крови к
свертыванию, появляются множественные кровоизлияния (геморрагический синдром). С
аутоинтоксикацией связано поражение почек, развитие печеночно-почечной недостаточности.
Гематоидин - не содержащий железа пигмент, кристаллы которого имеют вид ярко-
оранжевых ромбических пластинок или иголок, реже - зерен. Он возникает при распаде
эритроцитов и гемоглобина внутриклеточно, но в отличие от гемосидерина в клетках не
остается и при гибели их оказывается свободно лежащим среди некротических масс.
Химически он идентичен билирубину.
Скопления гематоидина находят в старых гематомах, рубцующихся инфарктах, причем в
центральных участках кровоизлияний - вдали от живых тканей.
Гематины представляют собой окисленную форму гема и образуются при гидролизе
оксигемоглобина. Они имеют вид темно-коричневых или черных ромбовидных кристаллов или
зерен, дают двойное лучепреломление в поляризованном свете (анизотропны), содержат
железо, но в связанном состоянии.
К выявляемым в тканях гематинам относят: гемомеланин (малярийный пигмент),
солянокислый гематин (гемин) и формалиновый пигмент. Гистохимические свойства этих
пигментов идентичны.
Гемомеланин (малярийный пигмент) возникает из простетической части гемоглобина под
влиянием плазмодиев малярии, паразитирующих в эритроцитах. При малярии
развивается гемомеланоз, так как при разрушении эритроцитов малярийный пигмент
попадает в кровь и подвергается фагоцитозу макрофагами селезенки, печени, костного мозга,
96
лимфатических узлов, головного мозга (при малярийной коме). Эти органы приобретают
аспидно-серую окраску. В них наряду с малярийным пигментом наблюдается отложение
гемосидерина.
Солянокислый гематин (гемин) находят в эрозиях и язвах желудка, где он возникает под
воздействием на гемоглобин ферментов желудочного сока и хлористоводородной кислоты.
Область дефекта слизистой оболочки желудка приобретает буро-черный цвет.
Формалиновый пигмент в виде темно-коричневых игл или гранул встречается в тканях при
фиксации их в кислом формалине (этот пигмент не образуется, если формалин имеет рН
>6,0). Его считают производным гематина.
Порфирины - предшественники простетической части гемоглобина, имеющие, как и гем, то
же тетрапиррольное кольцо, но лишенное железа. По химической природе порфирины близки
билирубину: они растворимы в хлороформе, эфире, пиридине. Метод выявления порфиринов
основан на способности растворов этих пигментов давать красную или оранжевую
флюоресценцию в ультрафиолетовом свете (флюоресцирующие пигменты). В норме
порфирины обнаруживаются в крови, моче, тканях. Они обладают свойством повышать
чувствительность организма, прежде всего кожи, к свету и являются поэтому антагонистами
меланина.
При нарушениях обмена порфиринов возникают порфирии, для которых характерно
увеличение содержания пигментов в крови (порфиринемия) и моче (порфиринурия), резкое
повышение чувствительности к ультрафиолетовым лучам (светобоязнь, эритема, дерматит).
Различают приобретенную и врожденную порфирии.
Приобретенная порфирия наблюдается при интоксикациях (свинец, сульфазол,
барбитураты), авитаминозах (пеллагра), пернициозной анемии, некоторых заболеваниях
печени. Отмечаются нарушения функции нервной системы, повышенная чувствительность к
свету, нередко развиваются желтуха, пигментация кожи, в моче обнаруживают большое
количество порфиринов.
Врожденная порфирия - редкое наследственное заболевание. При нарушении синтеза
порфирина в эритробластах (недостаточность уропорфириногена III - косинтетазы)
развивается эритропоэтическая форма,
а при нарушении синтеза порфирина в клетках печени (недостаточность уропорфирина III -
косинтетазы) - печеночная форма порфирии. При эритропоэтической форме порфирии
развивается гемолитическая анемия, поражаются нервная система и желудочно-кишечный
тракт (рвота, диарея). Порфирины накапливаются в селезенке, костях и зубах, которые
приобретают коричневый цвет; моча, содержащая большое количество порфиринов,
становится желто-красной. При печеночной форме порфирии печень увеличивается,
становится серо-коричневой, в ожиревших гепатоцитах, помимо отложений порфиринов,
находят гемосидерин.
97
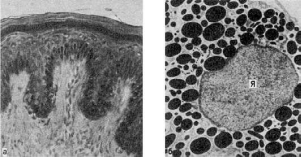
Нарушения обмена протеиногенных (тирозиногенных) пигментов
К протеиногенным (тирозиногенным) пигментам относят меланин, пигмент гранул
энтерохромаффинных клеток и адренохром. Накопление этих пигментов в тканях служит
проявлением ряда заболеваний.
Меланин (от греч. melas - черный) - широко распространенный буро-черный пигмент, с
которым у человека связана окраска кожи, волос, глаз. Он дает положительную
аргентаффинную реакцию, т.е. обладает способностью восстанавливать аммиачный раствор
нитрата серебра до металлического серебра. Эти реакции позволяют гистохимически
отличить его в тканях от других пигментов.
Синтез меланина происходит из тирозина в клетках меланинобразующей ткани -
меланоцитах, имеющих нейроэктодермальное происхождение. Их предшественниками
являются меланобласты. Под действием тирозиназы в меланосомах меланоцитов (рис. 39) из
тирозина образуется диоксифенилаланин (ДОФА), или промеланин, который полимеризуется
в меланин. Клетки, фагоцитирующие меланин, называютмеланофагами.
Рис.