

Гематологические заболевания, сопровождающиеся увеличением лимфоузлов злокачественные лимфомы
Под термином «злокачественные лимфомы», или «гематосаркомы», объединяют заболевания, характеризующиеся первичным локальным злокачественным опухолевым ростом, исходящим из лимфатической ткани.
В группе злокачественных лимфом выделяют:
1. Лимфогранулематоз (болезнь Ходжкина), который характеризуется гранулематозными разрастаниями с наличием клеток Березовского-Штернберга.
2. Неходжкинские лимфомы, для которых свойственна нодулярная или диффузная пролиферация, как правило, мономорфных патологических клеточных элементов.
Болезнь Ходжкина (лимфогранулематоз, лимфома Ходжкина)
Лимфогранулематоз (ЛГМ) – это опухолевое заболевание, при котором первично поражается лимфатическая система. Это заболевание неизвестной этиологии со злокачественными клетками Рида-Штернберга (в отечественной литературе названных клетками Березовского-Штернберга). Приблизительно у 25 % пациентов в ядрах клеток Рида-Штернберга был найден вирус Эпштейна-Барра, но значение этого феномена неизвестно, и происхождение болезни остается в значительной степени невыясненным.
Болеют лимфогранулематозом люди любого возраста, и кривая заболеваемости имеет два пика. Первый пик приходится на возраст 15-40 лет, достигая максимума в интервале 20-25 лет. Затем, после значительного снижения, кривая заболеваемости вновь начинает возрастать после 50 лет.
В основе ЛГМ лежит образование полиморфноклеточных гранулем с разрастанием фиброзных структур в лимфатических узлах и органах.
Гранулема состоит из:
- лимфоцитов,
- ретикулярных клеток,
- нейтрофилов,
- эозинофилов,
- плазматических клеток,
- клеток Рида-Штернберга (это крупные клетки d = 25-80 мкм, с базофильной цитоплазмой, содержащей 2 и больше круглых или овальных ядра),
- клеток Ходжкина (это крупные одноядерные клетки с базофильной цитоплазмой, ядро которых сходно с ядрами клеток Рида-Штернберга).
Клетки Рида-Штернберга и Ходжкина в 80% случаев происходят из зрелых, медленно пролиферирующих В-лимфоцитов терминального (зародышевого) центра фолликулов лимфатического узла, а в 20% являются дериватами (потомками) Т-клеточной линии цитотоксических лимфоцитов и, что менее вероятно, – дериватами натуральных киллеров (NK клеток). В ряде исследований было установлено, что большинство лимфоцитов, окружающих клетки Ходжкина и Рид-Штернберга, несут на своей поверхности рецепторы CD15 и CD30, т.е. являются опухолевыми клетками. Многоядерность клеток Рид-Штернберга объясняется нарушениями в центромере, которые возникают в результате соматической мутации и приводят к делению ядра, но последующего деления клетки не наступает. В большинстве случаев клетки Ходжкина и Рид-Штернберга являются результатом моноклональной пролиферации зрелых В-лимфоцитов терминального центра лимфатического узла, которые утратили способность экспрессировать иммуноглобулин и, избежав апоптоза, получили возможность неконтролируемой пролиферации (рис. 6).
Лимфогранулематоз сопровождается угнетением Т-клеточного иммунитета. Наиболее часто больные лимфогранулематозом подвержены вирусным инфекциям, в первую очередь герпетическим. У 16 % развивается herpes zoster, который протекает гораздо тяжелее, чем у здоровых людей, часто рецидивирует и при прогрессировании лимфогранулематоза имеет тенденцию к развитию некротических форм и к генерализации. Нередко наблюдается сочетание лимфогранулематоза с туберкулезом (в 5 % случаев).
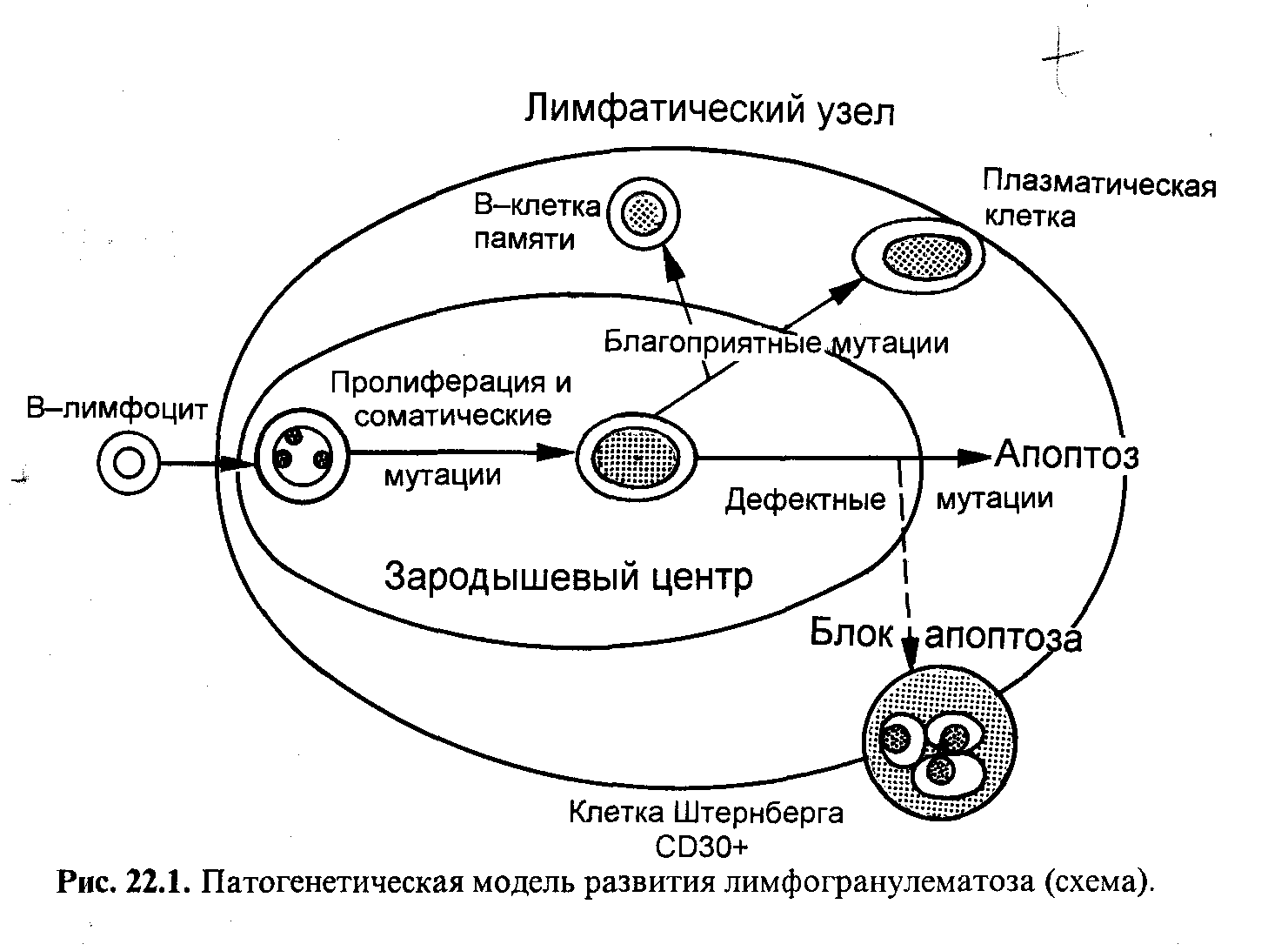
Рис. 6. Патогенетическая модель развития лимфогранулематоза
Существуют четыре гистологических варианта ЛГМ (классификация была предложена в 1966 г. R.J.Lukes, J.J.Butler, E.D.Hicks): с преобладанием лимфоцитов, нодулярный склероз, смешанно-клеточный и с истощением лимфоцитов (таб. 2).
Таблица 2. Гистологическая классификация лимфогранулематоза
|
Гистологический вариант
|
Морфологическая характеристика
|
Частота, % |
Средняя 5- летняя вы-живаемость, % |
|
Лимфогистиоцтарный (с преобладанием лим-фоцитов) |
Пролиферация лимфоцитов (диффузная или очаговая), увеличенное количество гистиоцитов, единичные гранулоциты и плазматические клетки, немногочисленные клетки Березовского-Штернберга, часто встречаются клетки Ходжкина; фиброза и некрозов нет |
3-5 |
90 |
|
Смешанноклеточный тип |
Выраженный клеточный полиморфизм, инфильтрат состоит из лимфоцитов, плазматических клеток, нейтрофилов, эозинофилов, большого количества клеток Березовского-Штернберга; часто обнаруживаются поля фиброза и очаги некроза |
25 |
35 |
|
Нодулярный склероз (узелковосклеротический тип) |
Узелки лимфомы Ходжкина окружены плотной фиброзной тканью с двойным лучепреломлением в поляризованном свете, тяжи коллагена располагаются концентрически; имеются клетки Березовского-Штернберга, зозинофилы, нейрофилы, лимфоциты, плазматические клетки; встречаются «лакунарные» клетки с широкой «пустой» цитоплазмой |
65-67 |
70 |
|
Лимфоцитопенический («лимфоидное истощение») |
Резкое уменьшение количества лимфоцитов; разрастание фиброзной ткани; многочисленные клетки Березовского-Штернберга; часто встречаются клетки Ходжкина |
5 |
35 |
В последней Европейско-американской классификации лимфоидных опухолей (Revised Еuropean American Lymphoma − REAL, 1994) классификация лимфогранулематоза почти не изменена. Выделен только условный пятый вариант «богатая лимфоцитами классическая болезнь Ходжкина». В марте 1998 г. на Четвертом международном симпозиуме по лимфогранулематозу в Кельне было решено изменить название «болезнь Ходжкина» (Hodgkin's disease) на «лимфому Ходжкина» (Hodkin's lymphoma).
Варианты: нодулярный склероз, смешанно-клеточный, лимфоидное истощение и условный вариант «богатая лимфоцитами классическая болезнь Ходжкина» имеют одинаковый иммунологический фенотип и экспрессируют сигнальные для лимфогранулематоза маркеры CD15 и CD30. Эти варианты объединены термином «классическая форма болезни Ходжкина». Вариант лимфоидное преобладание по клиническим, морфологическим и иммунофенотипическим характеристикам отличается от классической формы болезни Ходжкина и имеет совсем иной иммунологический фенотип и морфологические особенности.
Подразделение на варианты имеет большое прогностическое значение. Установлено, что прогностически благоприятными являются лимфоидное преобладание и нодулярный склероз, а неблагоприятными – смешанно-клеточный и лимфоидное истощение.
Клинические особенности и лечение.
ЛГМ обычно проявляется поражением шейных, надключичных или медиастинальных лимфатических узлов. Реже первым вовлеченным участком являются тазовые лимфатические узлы. ЛГМ постепенно распространяется на лимфатические узлы смежных областей. В конечном счете, поражаются печень, селезёнка, костный мозг и другие экстранодальные зоны. Возможно бессимптомное увеличение лимфатических узлов шеи или наличие симптомов, связанных с медиастинальной аденопатией. При прогрессировании болезни развиваются так называемые В-симптомы, включающие лихорадку, ночные поты и потерю веса.
Диагностические критерии лимфогранулематоза
Локальное (чаще шейных) или генерализованное увеличение лимфатических узлов: лимфоузлы плотные, безболезненные, не спаяны с кожей (при переходе процесса на капсулу лимфоузлы постепенно спаиваются в массивные конгломераты).
Лихорадка неправильного, волнообразного или постоянного характера (постоянная лихорадка наблюдается при далеко зашедшем процессе).
Проливной пот.
Кожный зуд (у 25-40 % больных зуд генерализованный или локальный – в области увеличенных лимфоузлов).
Сплено- и гепатомегалия.
Изменения гемограммы: лейкоцитоз (реже – лейкопения) с нейтрофилёзом и лимфопенией, выраженное увеличение СОЭ.
Определение в биоптате узла или селезёнки клеток Березовского-Штернберга и одного из морфологических вариантов заболевания – лимфогистиоцитарного, нодулярного склероза, смешанно-клеточного, лимфоидного истощения (обязательный критерий).
Диагноз лимфогранулематоза устанавливается исключительно морфологически и считается доказанным только в том случае, если при гистологическом исследовании найдены специфические многоядерные клетки Штернберга.
Когда при биопсии подтверждается диагноз ЛГМ, необходимо установить стадию болезни на основании данных КТ-сканирования груди, живота и таза, а также биопсии костного мозга. По Международной клинической классификации (Ann-Arbor, 1971 г.) лимфогранулематоз делится на четыре стадии (табл. 3).
Таблица3. Стадии лимфогранулематоза
|
Стадия I |
Поражение одной лимфатической зоны или структуры (I) или локализованное поражение одного экстралимфатического органа или ткани (IE) в пределах одного сегмента. |
|
Стадия II |
Поражение двух или более лимфатических областей по одну сторону диафрагмы (например, средостение – одна зона, корни легких – отдельные от средостения самостоятельные зоны) либо локализованное поражение одного экстралимфатического органа или ткани и их регионарных лимфатических узлов с (или без) поражением других лимфатических областей по ту же сторону диафрагмы или без него (II Е). Для II стадии следует указывать число пораженных лимфатических зон, например II4. |
|
Стадия III |
Поражение лимфатических узлов или структур по обе стороны диафрагмы, которое может сочетаться с локализованным поражением одного экстралимфатического органа или ткани (IIIE) либо с поражением селезенки (IIIS) или поражением того и другого (IIIE+S). Рекомендуется выделять верхние абдоминальные лимфатические узлы (ворота печени, селезенки, чревные) – стадия III1 и нижние абдоминальные (парааортальные, мезентериальные) стадия III2. |
|
Стадия IV |
Диссеминированное (многофокусное) поражение одного или нескольких экстралимфатических органов, с поражением лимфатических узлов или без него, либо изолированное поражение экстралимфатического органа с поражением отдаленных (не регионарных) лимфатических узлов. Метастазы в печень и костный мозг – всегда IV стадия. |
Поражение селезенки обозначается символом «S» (стадии IS, IIS, IIIS). Символом «Е» обозначается локализованное (в пределах одного сегмента) экстранодальное поражение (стадии IE, IIЕ, IIIE). Символом «X» предложено обозначать массивное поражение лимфатических узлов. Отдельно обозначают симптомы интоксикации: символом «В» – наличие одного или более из следующих клинических симптомов: ночной профузный пот, температура тела выше 38 °С не менее 3 дней подряд без признаков воспалительного процесса, снижение массы тела на 10 % за последние 6 месяцев; символом «А» – отсутствие указанных выше симптомов. Кожный зуд исключен из симптомов интоксикации.
Кроме стадии и симптомов интоксикации, для выбора тактики и объема лечения больных лимфогранулематозом Германская группа по изучению лимфомы Ходжкина рекомендует использовать группу прогностических факторов, так называемые «факторы риска», которые в большей или меньшей степени определяют прогноз заболевания. К ним относят:
1) экстранодальное поражение в пределах, обозначаемых символом Е;
2) расширение тени средостения на рентгенограммах увеличенными лимфатическими узлами более чем на 1/3 диаметра грудной клетки в самом широком ее месте;
3) массивное поражение селезенки (наличие 5 или более очагов или диффузное поражение увеличенного органа);
4) поражение 3 или более зон лимфатических узлов;
5) ускорение СОЭ выше 30 мм/ч в стадии В и выше 50 мм/ч в стадии А.
В соответствии с объёмом опухолевой массы выделены три прогностические группы:
1) больные с благоприятным прогнозом (больные в I стадии и во II стадии без факторов риска);
2) промежуточным прогнозом (больные IIВ и факторами риска 4 и 5, больные в III1А стадии без факторов риска);
3) неблагоприятным прогнозом (больные в II стадии и факторами риска 1-3, больные в IIIА с любыми факторами риска и больные в IIIВ и IV).
План обследования.
1) Сбор анамнеза для уточнения наличия у больного симптомов интоксикации (повышение температуры тела, профузный пот, похудание).
2) Тщательный осмотр больного, включая: пальпацию всех периферических лимфатических узлов (в том числе подключичных, локтевых и подколенных), пальпацию брюшной полости, в том числе печени, селезенки и области забрюшинных и подвздошных лимфатических узлов.
3) Пункцию, а затем обязательную адекватную биопсию лимфатического узла. При изолированном увеличении медиастинальных или внутрибрюшных лимфатических узлов следует прибегнуть к парастернальной медиастинотомии или лапаротомии.
4) Из лабораторных тестов обязательными являются:
а) полный клинический анализ крови;
б) биохимические пробы − исследование уровня щелочной фосфатазы, функции печени и почек, а также по возможности определение признаков биологической активности (содержание α2-глобулина, фибриногена, гаптоглобина и церулоплазмина).
5) Всем больным показано рентгенографическое исследование грудной клетки в прямой и боковой проекциях. В отсутствие изменений на рентгенограммах обязательна компьютерная томография грудной клетки, которая позволяет выявить невидимые на стандартных рентгенограммах медиастинальные лимфатические узлы и мелкие очаги в легочной ткани.
6) Ультразвуковое исследование печени, селезенки, забрюшинных и внутрибрюшных лимфатических узлов, почек помогает исключить (или подтвердить) поражение этих органов. В сомнительных случаях показана компьютерная томография брюшной полости.
7) Методы радиоизотопной диагностики позволяют выявить субклиническое поражение костной системы. Все зоны патологического накопления индикатора, особенно если по локализации они совпадают с болями в костях, на которые жалуется больной, следует подтвердить рентгенографически.
8) Трепанобиопсия подвздошной кости – обязательная процедура, так как является единственным методом, подтверждающим или исключающим специфическое поражение костного мозга.
При подозрении на поражение других органов необходимо обследование соответствующими методами.
Лечение
Современное индукционное (первичное) лечение приводит к полной ремиссии у 60–90 % первичных больных лимфогранулематозом, а 20-летняя безрецидивная выживаемость в группе больных с полной ремиссией после лечения по первой программе превышает 60 %.
МОПП (и ее модификации) и схема ABVD и сегодня являются основными программами первой линии для больных лимфогранулематозом.
Схемы первой линии:
МОРР (МОПП):
мустарген (эмбихин) - 6 мг/м2 внутривенно в 1-й и 8-й дни
онковин (винкристин) - 1,4 мг/м2 (максимум 2 мг) в/в в 1-й и 8-й дни
прокарбазин (натулан) - 100 мг/м2 внутрь ежедневно с 1-го по 14-й день преднизолон - 40 мг/м2 внутрь ежедневно с 1-го по 14-й день.
Перерыв между циклами 2 недели.
MVPP (МВПП):
мустарген (эмбихин) - 6 мг/м2 внутривенно в 1-й и 8-й дни
винбластин (розевин) - 6 мг/м2 внутривенно в 1-й и 8-й дни
прокарбазин (натулан) - 100 мг/м2 внутрь ежедневно с 1-го по 14-й день
преднизолон - 40 мг/м2 внутрь ежедневно с 1-го по 14-й день.
Перерыв между циклами 3-4 недели.
CVPP (ЦВПП):
циклофосфан - 650 мг/м2 внутривенно в 1-й и 8-й дни
винбластин (розевин) - 6 мг/м2 внутривенно в 1-й и 8-й дни
прокарбазин (натулан) - 100 мг/м2 внутрь ежедневно с 1-го по 14-й день
преднизолон - 40 мг/м2 внутрь ежедневно с 1-го по 14-й день в
1-м и 4-м циклах.
Перерыв между циклами 2 недели.
Лечение больных во II стадии без неблагоприятных факторов: 2 цикла полихимиотерапии по схеме ЦВПП + облучение только зон исходного поражения в СОД (суммарная очаговая доза), не превышающей 36-40 Гр + 2 цикла ЦВПП. Такая программа лечения позволяет получить 98 % полных ремиссий, 6-летнее безрецидивное течение составляет 94 %, а 6-летняя общая выживаемость – 100 % .
Лечение больных во II стадии с факторами риска (при наличии хотя бы одного из них): 3 цикла полихимиотерапии по одной из схем первой линии + облучение всех лимфатических коллекторов выше диафрагмы в СОД до 40 Гр + 3 цикла такой же полихимиотерапии, как и вначале. Такая программа лечения позволяет получить 96-98 % полных ремиссий при 79-90 % 6-летней безрецидивной выживаемости и 85-91 % 6-летней общей выживаемости.
В последнее десятилетие наметилась тенденция к уменьшению объема лучевой терапии и суммарных очаговых доз в комбинированных химиолучевых программах. Больных во IIB стадии с большим средостением, экстранодальным поражением в пределах стадии Е, в IIIA стадии и с факторами риска, в IIIB и IV стадиях предложено объединить в группу с неблагоприятным прогнозом. Этим больным показана более интенсивная терапия:
Программа VD (VELP-DCHOP):
этопозид - 300 мг/м2 внутривенно в 1-й день
винбластин - 6 мг/м2 внутривенно в 1-й день
хлорамбуцил - 6 мг/м2 внутрь ежедневно в 1—10-й дни
преднизолон - 40 мг/м2 внутрь ежедневно в 1—10-й дни
дакарбазин - 500 мг/м2 внутривенно на 21-й день
адриамицин - 50 мг/м2 внутривенно на 21-й день
циклофосфан - 600 мг/м2 внутривенно на 21-й день
винкристин - 1,4 мг/м2 внутривенно на 21-й день
преднизолон - 40 мг/м2 внутрь ежедневно в 21—30-й дни.
Курс повторяют на 42-й день. Лечение проводят до полной ремиссии + 5 консолидирующих курсов, но в сумме не более 10 + лучевая терапия в суммарной очаговой дозе 30 Гр на зоны массивных исходных поражений.
При лечении по программе VD (VELP-DCHOP) полные ремиссии получены у 96 % больных при медиане без рецидивного течения 71 мес. и медиане общей выживаемости 78 мес.
Для подтверждения ремиссии необходимо исследование крови, стандартное рентгенологическое исследование, ультразвуковая томография всех зон исходного поражения, компьютерная томография грудной клетки и при необходимости брюшной полости, а у больных с исходно большими размерами средостения — и сканирование лимфатических узлов с 67Ga, которое успешнее всего используется при выявлении резидуальных лимфатических узлов в средостении. Кроме того, при поражении костей, костного мозга и других органов по окончании лечения должны быть проконтролированы все эти зоны исходного поражения.
Больных с рецидивами после полихимиотерапии и или индукционного комбинированного химиолучевого лечения по характеру течения ЛГМ и ответу на повторное лечение можно разделить на 3 группы:
Больные, не достигшие полной ремиссии после первой программы лечения (первично-резистентные).
Больные, у которых ремиссия сохранялась менее 1 года.
Больные, у которых ремиссия сохранялась более 1 года.
При лечении повторных рецидивов в зависимости от времени их возникновения используют схемы, как первой, так и второй линии. Однако каждый следующий рецидив снижает шансы больного на долговременную выживаемость: 10-летний рубеж переживают менее 10% больных с 4-м рецидивом.
Неходжкинские лимфомы
Неходжкинские лимфомы (НХЛ) — это гетерогенная группа злокачественных лимфопролиферативных опухолей, различающихся по биологическим свойствам, морфологическому строению, клиническим проявлениям, ответу на терапию и прогнозу. Все они характеризуются клональным размножением, пролиферациями лимфоидных клеток типа В или Т.
Возраст является наиболее значимым фактором риска развития НХЛ: уровень заболеваемости в возрасте 15-20 лет более чем в 10 раз ниже по сравнению с таковым в возрасте старше 75 лет (эта особенность характерна для обоих полов).
Этиология большинства случаев неходжкинских лимфом неизвестна. Аномальные лимфоидные клетки способны циркулировать в периферической крови; общим признаком неходжкинских лимфом является гематогенная диссеминация. Скорость прогрессирования (роста) среди различных типов лимфом вариабельна. Некоторые лимфомы медленно прогрессируют, а другие растут чрезвычайно быстро.
Классификация НХЛ представлена в таблице 4.
Таблица 4. Современная Кильская классификация лимфом
(модифицирована в 1992 г.)
|
В-лимфомы |
Т-лимфомы |
|
Лимфомы низкой степени злокачественности | |
|
Лимфоцитарные: хронический лимфолейкоз пролимфоцитарный лейкоз волосато-клеточный лейкоз
Лимфоплазмоцитарная / цитоидная (иммуноцитома) Плазмоцитарная Центробластная-центроцитарная: фолликулярная±диффузная диффузная Центроцитарная (из клеток мантии) Моноцитойдная (в том числе из клеток маргинальной зоны) |
Лимфоцитарные: хронический лимфолейкоз пролимфоцитарный лейкоз
Из мелких церебриформных клеток: грибовидный микоз, синдром Сезари Лимфоэпителиоидная (лимфома Леннерта)
Ангиоиммунобластная (AILD, LgX) Лимфома Т-зоны
Плеоморфная из мелких клеток (HTLV±)
|
|
Лимфомы высокой степени злокачественности | |
|
Центробластная
Иммунобластная Лимфома Беркитта Крупноклеточная анапластическая (Ki-l+) Лимфобластная Редкие типы |
Плеоморфная из средних и крупных клеток (HTLV±) Иммунобластная (HTLV±)
Крупноклеточная анапластическая (Ki-l+) Лимфобластная Редкие и неклассифицируемые типы |
В Кильской классификации отсутствуют иммунологические критерии выделения вариантов в пределах В- и Т-клеточных лимфом (см. Приложение 1). Данный пробел в значительной степени восполнен в REAL – классификации (уточнённая европейско-американская классификация лимфом), которая остаётся в большей степени морфологической, чем иммунологической.
Пересмотренная классификация неходжкинских лимфом (REAL)
В-клеточные
1. НХЛ из предшественников В-клеток
- пре-В-клеточный лимфобластный лейкоз/лимфома
2. В-клеточные опухоли из клеток с «периферическим» фенотипом (зрелых)
а. В-клеточный ХЛЛ/мелкоклеточная лимфоцитарная лимфома
б. Лимфоплазмоцитоидная лимфома/иммуноцитома
в. Лимфома из мантийных клеток
г. Лимфома из клеток фолликулярных центров, фолликулярная
- из малых клеток, смешанная из малых и больших клеток,
из больших клеток
- диффузная, преимущественно из малых клеток
д. В-клеточная НХЛ маргинальной зоны
- экстранодальная (MALT ± моноцитоидная)
- нодальная (± моноцитоидная)
- селезеночная (± ворсинчатые лимфоциты)
е. Волосатоклеточный лейкоз
ж. Плазмоцитома/миелома
з. Диффузная крупноклеточная В-клеточная лимфома
подтип: первичная медиастинальная В-клеточная
и. Лимфома Беркитта
Т-клеточные
1. НХЛ из предшественников Т-клеток
а. Т-лимфобластная лимфома/лейкоз
2. Т-клеточная НХЛ из клеток с «периферическим» фенотипом и NK-клеточные опухоли
а. Т-клеточный ХЛЛ/пролимфоцитарный лейкоз
б. Крупногранулярный лимфоцитарный лейкоз (Т-клеточный и NK-клеточный)
в. Грибовидный микоз/синдром Сезари
г. Т-клеточная лимфома с «периферическим» фенотипом
- смешанная НХЛ из средних и больших клеток, из больших клеток, лимфоэпителиоидная
д. Ангиоиммунобластная Т-клеточная лимфома (AILD)
е. Ангиоцентрическая лимфома
ж. Кишечная Т-клеточная лимфома (± энтеропатия)
з. Т-клеточная лимфома/лейкоз взрослых (HTLV-1)
и. Анапластическая крупноклеточная лимфома (CD30+), Т- и нулевого клеточного типа
Опухоли низкой степени злокачественности характеризуются медленным прогрессированием, длительной выживаемостью (годы), умеренной чувствительностью к химиотерапии и отсутствием возможности излечения при стандартной химиотерапии. В эту группу включены следующие варианты НХЛ (в соответствии с классификацией REAL):
В-клеточные опухоли.
1. Фолликулярная НХЛ (I−II степени).
2. Диффузная лимфоцитарная НХЛ.
3. НХЛ маргинальной зоны: а) malt-экстраодалъная; б) моноцитовдная − нодальная; в) НХЛ селезенки.
Т-клеточные НХЛ.
Грибовидный микоз/синдром Сезари.
Значительных различий в выживаемости внутри этой группы не выявлено.
Высокоагрессивные лимфомы быстро прогрессируют, выживаемость составляет лишь месяцы, они умеренно и высокочувствительны к химиотерапии и могут быть излечены стандартными методами химиотерапии. Однако внутри этой группы НХЛ выявляются весьма значимые различия в продолжительности жизни: 5-летняя выживаемость колеблется от 78 % при анапластической крупноклеточной лимфоме до 14 % при лимфоме зоны мантии, в то время как при диффузной В-крупноклеточной лимфоме и фолликулярной лимфоме III степени этот показатель равен 38 и 68 % соответственно. В эту группу включены следующие варианты:
В-клеточные опухоли.
1. Диффузная крупноклеточная НХЛ.
2. НХЛ Беркитта и беркиттоподобные опухоли.
Т-клеточные НХЛ.
1. Лимфобластная лейкемия/лимфома.
2. Периферические Т-клеточные НХЛ.
3. Анапластическая крупноклеточная НХЛ.
4. Ангиоиммунобластная НХЛ.
СТАДИИ ЛИМФОМ
Стадия I:
Вовлечение одной группы лимфатических узлов с любой стороны диафрагмы; непосредственное поражение ограниченной области или наличие одного экстранодального очага, являющегося единственным проявлением заболевания.
Стадия II:
Вовлечение 2-х и более групп лимфатических узлов по одну сторону диафрагмы; может вовлекаться селезенка, если некоторые группы лимфатических узлов расположены ниже диафрагмы.
Стадия Ш:
Вовлечение групп лимфатических узлов с обеих сторон диафрагмы, может вовлекаться селезенка.
Стадия IV:
Вовлечение экстранодальных участков, таких, как костный мозг или печень.
А: Отсутствие симптомов интоксикации
В: Лихорадка, ночные поты, потеря веса (более 10% массы тела) ±
кожный зуд.
Клиническая картина. НХЛ начинаются с появления одиночного опухолевого узла и распространяются путем лимфогенного и гематогенного метастазирования. Первичный опухолевый очаг может локализоваться в лимфатических узлах (нодальное поражение) или в других органах и тканях (экстранодальное поражение). Клинические проявления обусловлены расположением опухолевых очагов. Наиболее часто первыми проявлениями болезни бывает поражение лимфатических узлов (45-50 %); периферические лимфатические узлы вовлекаются в процесс значительно чаще (35-38 %), чем медиастинальные, забрюшинные и внутрибрюшные. Вначале лимфатические узлы плотны, безболезненны, не спаяны с кожей и подлежащими тканями. Позднее они образуют конгломераты, нередко довольно больших размеров. Изъязвление и образование свищей наблюдается не часто. При формировании свищевых ходов необходимо исключить туберкулез и актиномикоз.
При поражении лимфатических узлов шеи и средостения может наблюдаться сдавление пищевода и трахеи, вызывающее дискомфорт при глотании пищи и кашель. Сдавление крупных сосудов грудной полости (наиболее часто в заднем медиастинальном пространстве) обусловливает застой в системе верхней полой вены, проявляющийся цианозом и отечностью верхней половины тела и лица с нарушениями дыхания и тахикардией. Лимфатические узлы брюшной полости и забрюшинного пространства, достигая значительных размеров или локализуясь в функционально ответственных зонах, могут обусловить развитие кишечной непроходимости, застой в портальной системе и нарушение лимфооттока из нижней половины тела (как следствие – развитие асцита, отеков нижних конечностей, половых органов), появление механической желтухи, нарушение мочеотделения.
Опухолевые образования в носоглотке бугристые, бледно-розового цвета, быстро растут, могут прорастать решетчатый лабиринт, верхнечелюстную пазуху, глазницу, вызывая экзофтальм. Основными клиническими симптомами являются затруднение носового дыхания и снижение слуха. Глоточные миндалины могут значительно увеличиваться, а иногда (при двустороннем поражении) почти смыкаться между собой и быстро изъязвляться.
При поражении молочной железы опухоль может определяться в виде диффузного уплотнения или довольно четкого опухолевого узла с прорастанием кожи и подлежащих тканей или без него. Симптом умбиликации (втяжение соска) не выражен. Поражение яичка начинается с отдельного участка, но быстро распространяется на весь орган, как правило, не вызывая спонтанных болей и пальпация безболезненна, но иногда опухоль прорастает кожу мошонки и распадается с образованием язвенной поверхности. При сочетании поражения яичка с регионарными (пахово-подвздошными) лимфатическими узлами отмечается отек мошонки.
Лимфома кожи проявляется по-разному. Может иметь место постепенно растущий солитарный опухолевый узел в толще кожи и подкожной жировой клетчатке или появление вслед за маленьким внутрикожным узелком множества других узлов разных размеров. Обычно они плотны, безболезненны, могут располагаться группами и сливаться в плотные инфильтраты с бугристой поверхностью, склонные к изъязвлению. Цвет кожи над ними может быть не изменен или она приобретает темно-багровую окраску. У части больных первичные проявления протекают по типу дерматитов.
Особенно характерная клиническая картина развивается при поражении ЦНС. Как правило, это является следствием гематогенного диссеминирования НХЛ и обнаруживается в среднем через 1,5 года от начала болезни преимущественно при НХЛ высокой степени злокачественности. Реже встречается изолированное первичное поражение ЦНС. Клиническая картина может быть мало выраженной и зависит от локализации опухоли: появление одиночного или реже множественных узловых образований в веществе головного мозга сопровождается неврологической симптоматикой поражения различных отделов головного мозга, а при вовлечении в процесс оболочек мозга наблюдается картина, напоминающая менингит (резкая головная боль, тошнота, рвота, положительные менингеальные симптомы).
При расположении опухоли в желудочно-кишечном тракте не наблюдается специфических, присущих только НХЛ клинических признаков. Больные предъявляют жалобы, которые отмечаются при любой опухоли этой локализации (тошнота, ухудшение аппетита, снижение массы тела). Выраженность клинических проявлений заболевания обусловлена локализацией и формой роста опухоли. Наиболее часто поражаются желудок и тонкая кишка, как при изолированном, так и при сочетанием вовлечении их в процесс.
Клинический вариант заболевания с исходным локализованным поражением экстранодальных органов и тканей определяется как первичная экстранодальная НХЛ. Подобная трактовка клинической ситуации возможна при выявлении единственного экстранодального опухолевого очага или в сочетании с последующим вовлечением в процесс регионарных лимфатических узлов (клинические стадии IE или IIЕ).
Диагностика. Заболевание диагностируют на основании морфологического исследования опухолевого образования. Цитологическое исследование высокоинформативно. Следует широко выполнять его в амбулаторных условиях. Окончательным следует считать гистологическое исследование биоптата опухолевой ткани с иммунофенотипированием (Приложение 1). Цитологическая верификация допускается только в тех случаях, когда взятие материала для гистологического исследования cопряжено с высоким риском для жизни. Чаще всего выполняют биопсию периферических лимфатических узлов. Необходимо отдавать предпочтение биопсии длительно существующего лимфатического узла, расположенного в легко доступных анатомических областях (лучше шейно-надключичной области). Менее желательна биопсия паховых и апикальных аксиллярных лимфатических узлов).
Целью иммунофенотипирования является определение В- и Т-клеточного происхождения опухоли и уровня нарушения дифференцировки лимфоидной клетки. Этот метод являются высокоинформативным компонентом комплексной диагностики НХЛ.
Наиболее часто (более чем в 90 % случаев) НХЛ имеет В-клеточное происхождение, экспрессируя пан-В-клеточные антигены: CD19, CD20, CD22, обычно в сочетании с HLA/DR и молекулами поверхностных иммуноглобулинов. Наличие других В-клеточных антигенов (CD5, CD10, CD38, CD23 и др.) позволяет с наибольшей достоверностью установить В-клеточный вариант НХЛ, что лежит в основе выбора адекватной лечебной тактики. Для Т-клеточных опухолей характерно наличие CD4, CD7, CD8.
Поскольку возможно расположение опухолей в любых органах и тканях, план обследования должен включать: полный физикальный осмотр с исследованием всех групп периферических лимфатических узлов, рентгенологическое исследование органов грудной клетки (для определения состояния медиастинальных лимфатических узлов и легочной ткани), ультразвуковое исследование печени, селезенки, внутрибрюшных и забрюшинных лимфатических узлов, фиброларингоскопию для выяснения состояния лимфа-тического аппарата кольца Пирогова-Вальдейера, рентгенологическое исследование желудка или, предпочтительно, гастроскопию с биопсией суспициозных участков слизистой оболочки, стернальную пункцию и трепанобиопсию подвздошной кости для исключения лейкемической трансформации и гематогенного очагового поражения костного мозга. Кроме того, нужно исследовать органы, со стороны которых больной испытывает дискомфорт.
При особых клинических вариантах НХЛ применяют специфические дополнительные методы обследования: при поражении оболочек головного и спинного мозга (после осмотра неврологом) – люмбальную пункцию с определением клеточности цереброспинальной жидкости, и биохимического и цитологического исследования, при опухолевом поражении ЦНС – компьютерную томографию головного мозга и (или) уровня поражения спинного мозга, установленного при неврологическом осмотре; при первичном поражении одного из отделов желудочно-кишечного тракта – дополнительное исследование всех отделов желудочно-кишечного тракта.
Диагностические критерии.
Диагностику лимфом (лимфоцитом) облегчают следующие характерные особенности, присущие зрелоклеточным опухолям лимфатической системы:
• очаговый характер лимфоидной пролиферации, поражение преимущественно какого-либо одного органа;
• доброкачественное течение заболевания на протяжении 10-20 лет;
• частое (около 25%) перерождение в лимфосаркому (А. И. Воробьев, 2000), которая очень чувствительна к химиотерапии и облучению и дает полную ремиссию;
• невысокий лимфоцитов в периферической крови;
• высокая частота секреции моноклонального иммуноглобулина (чаще IgM);
• обнаружение в биоптатах лимфатических узлов пролиферации зрелоклеточных лимфоцитов.
Прогноз. На основании тщательного изучения всех проявлений НХЛ было установлено, что наиболее значимыми факторами неблагоприятного прогноза являются: возраст старше 60 лет, повышение уровня ЛДГ (в 2 раза и более), общее состояние больного, соответствующее 2-4-й степени (ECOG), III − IV стадия болезни, наличие более одного экстранодального очага поражения, вовлечение костного мозга. Это было положено в основу Международного прогностического индекса – МПИ (IPI). Размеры опухолевых масс не включены в МПИ, хотя в ряде клинических ситуаций, бесспорно, играют отрицательную роль.
На основании наличия одновременно того или иного количества неблагоприятных факторов прогноза различают четыре группы (степени) риска раннего прогрессирования болезни: низкую – отсутствие или присутствие лишь одного неблагоприятного признака, низкую/промежуточную – наличие двух факторов, промежуточную/высокую − присутствие трех и высокую − четырех неблагоприятных факторов
Лечение. При НХЛ используют все виды противоопухолевой терапии. Основными факторами, влияющими на выбор терапевтической такта являются, распространенность процесса (I−II или III−IV клиническая стадия), морфологический вариант опухоли, первичная или преимущественная локализация опухолевого поражения, факторы прогноза.
Единственным поистине объективным критерием эффективности терапии является подтверждение наступления молекулярной ремиссии – документированное использованием метода полимеразной цепной реакции, отсутствие моноклонального опухолевого клона и присущих ему молекулярных изменений. Эта оценка всё шире внедряется в мировую практику.
Хирургическое лечение применяют с наименьшей частотой. Основным показанием к нему являются первичные одиночные опухоли желудочно-кишечного тракта. Спленэктомию выполняют редко, обычно с целью коррекции гематологических показателей при выраженном гиперспленизме (что часто сочетается со специфическим поражением селезенки). Лучевая терапия является высокоэффективным методом лечения НХЛ, может использоваться на всех этапах болезни, хотя возможности ее неодинаковы. В качестве самостоятельного метода лечения ее применяют редко, но успешно сочетают с другими лечебными воздействиями, особенно химиотерапией. Большое распространение получила комбинированная (химиолучевая) терапия в начальных (I–II А) стадиях НХЛ.
Лечение в I–II стадии. Комбинированное лечение всегда следует начинать с химиотерапии, которая приводит к полным ремиссиям не менее чем у 70 % больных уже к моменту начала лучевого этапа лечения. Несмотря на это, лучевую терапию обязательно включают в комплекс лечебных мероприятий, так как она дает более стойкие местные лечебные результаты. Осуществляется комбинированное химиолучевое лечение в виде «сэндвича»: 2-3 цикла полихимиотерапии – лучевая терапия – 2-3 цикла полихимиотерапии.
В I стадии НХЛ низкой степени злокачественности до и после лучевой терапии проводятся 2 цикла полихимиотерапии по схеме СОР (циклофосфан, винкристин, преднизолон). Альтернативным вариантом лечения больных этой группы могут быть 3 цикла полихимиотерапии с последующей лучевой терапией без дополнительной химиотерапии после облучения, если к моменту начала лучевой терапии была достигнута полная ремиссия.
Больным с НХЛ высокой степени злокачественности в I стадии до начала лучевой терапии и после нее требуется провести по 3 цикла полихимиотерапии. В комбинацию химиопрепаратов должны быть включены антрациклины. Наиболее удобной и эффективной является схеме CHOP (циклофосфамид, доксорубицин, винкристин, преднизолон).
Во IIА стадии всем больным независимо от степени злокачественности следует проводить 6 циклов полихимиотерапии: 3 до и 3 после лучевой терапии. Облучению подлежат только исходные зоны поражения (традиционный режим гамма-терапии до суммарной очаговой дозы 32–36 Гр). Профилактическое облучение всех групп лимфатических узлов нецелесообразно. Частота рецидивов составляет 18–30 %, но в течение первого года они возникают менее чем у 1/3 больных.
Лечение в III–IV стадии. Комбинированная химиолучевая терапия в III стадии по результатам не превосходит обычную, а в IV стадии малоэффективна, так как облучение отдельных доступных зон носит паллиативный характер. Основным методом лечения распространенных форм НХЛ является химиотерапия, интенсивность которой зависит от степени злокачественности опухоли и клинических проявлений.
НХЛ низкой степени злокачественности являются медленно прогрессирующими опухолями, которые характеризуются своеобразными клиническими особенностями.
В последнее время все шире используется при лимфомах низкой степени злокачественности митоксантрон (новантрон) как компонент полихимиотерапии. В случаях благоприятного прогноза в качестве I линии терапии наибольшее распространение получила комбинация МСР (митоксантрон – 14 мг/м2 однократно в 1-й день, лейкеран – 6 мг/м2 с 1-го по 10-й день и преднизолон − 25 мг/м2 с 1-го по 10-й день). Этой комбинацией все чаще заменяют схему СОР. При неблагоприятном прогнозе на первом этапе индукционной терапии используют комбинацию MAP (митоксантрон, цитарабин, преднизолон), OPEN (винкристин, преднизолон, этопозид, новантрон), NOPP (новантрон, винкристин, натулан, преднизолон), MVLP (митоксантрон, тенипозид, лейкеран, преднизолон). Такое лечение проводят при всех вариантах НХЛ низкой степени злокачественности.
НХЛ высокой степени злокачественности. Клиническими особенностями НХЛ высокой степени злокачественности являются быстрый рост опухолевых образований и склонность к раннему прогрессированию. Первой линией терапии диссеминированных НХЛ является схема СHОР, признанная «золотым стандартом». В настоящее время в рандомизированных исследованиях показана возможность повышения эффективности схемы СНОР у первичных больных: полная ремиссия достигается в 75-86% случаев, что превышает результативность стандартной схемы более чем на 20%. Это достигается интенсификацией режима двумя путями: сокращением интервала между циклами до 10-14 дней и (или) увеличением дозы доксорубицина до 70-80 мг/м2. Подобная интенсификация стала возможной лишь при обязательном использовании колониестимулирующих факторов – G-CSF, GМ-CSF (Г-КСФ, ГМ-КСФ). Разработана 2-х недельная модификация СНОР для лиц пожилого возраста (старше 70 лет) с заменой адриамицина форморубицином.