


Следовательно, в реакции третьего порядка время полупревращения обратно пропорционально квадрату начальной концентрации вещества.
Реакции n-го порядка. При равных начальных концентрациях реагентов дифференциальное уравнение для скорости реакции n-го порядка записывают в виде
dC |
= knCn. |
(31) |
− dt |
Разделение переменных и интегрирование в пределах от С0 до С приводит к уравнению
|
1 |
− |
1 |
= (n−1)knt. |
(32) |
|||||
|
Cn−1 |
Cn−1 |
||||||||
0 |
|
|
|
|
|
|
||||
Время полупревращения для реакции n-го порядка равно: |
|
|||||||||
|
t1/2 = |
(n |
2n−1 −1 |
. |
(33) |
|||||
|
|
|
|
− |
1)k |
n |
Cn−1 |
|
|
|
|
|
|
|
|
|
0 |
|
|
||
1.4. Некоторые методы определения порядка реакции
Современные методы вычислительной математики (различные методы минимизации) позволяют с помощью ЭВМ по кинетическим кривым, полученным экспериментально, непосредственно найти порядок реакции по отдельным реагентам. На практике часто используют более простые методы определения порядка реакций по экспериментальным данным.
Метод подстановки. Метод позволяет установить соответствие опытных данных одному из интегральных кинетических уравнений для целочисленных порядков реакции (от нуля до трех). На практике используют алгебраический и графический варианты метода подстановки. Расчет константы скорости реакции выполняют последовательно для реакций различных порядков (от нуля до трех). Сохранение постоянства значений k при подстановке опытных значений концентрации и времени в очередное кинетическое уравнение (в пределах ошибки эксперимента) свидетельствует о правильности выбора порядка реакции. Если ни одно кинетическое уравнение не дает удовлетворительного результата, можно
16
сделать заключение о сложном характере реакции, порядок которой может быть дробным и отрицательным. В этом случае следует использовать другие способы определения порядка реакции.
Определение порядка реакции по начальным скоростям (метод Вант-Гоффа). В соответствии с основным постулатом хи-
мической кинетики начальную скорость реакции A+B → P можно записать в виде
r0 = kC0An C0Bm .
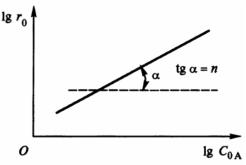
Для определения порядка реакции по веществу A проводят серию экспериментов при постоянной начальной концентрации вещества B (C0В = const) и различных начальных концентрациях вещества A (C0А = const). Тогда, логарифмируя уравнение для начальной скорости реакции, получаем
lgr0 = lgk +lgC0Bm +nlgC0A = lgk +nlgC0A, (34)
где k = kC0Bm . Зависимость lgr0 от lgC0А является прямой, тангенс угла наклона которой равен порядку реакции по веществу A (рис. 6).
Рис. 6. Графическое определение порядка реакции по начальным скоростям (С0В = const, С0А = const)
Аналогично по опытным данным, полученным в другой серии экспериментов, когда C0А = const, а C0В = const, определяют порядок реакции по веществу B. Значения скорости реакции находят графическим дифференцированием кинетической кривой.
17

Порядок реакции можно рассчитать аналитически, если известны начальные скорости реакции при двух исходных концентрациях вещества:
r01 = k1C0A1n ; r02 = k1C0A2n .
Возьмем отношение этих скоростей и прологарифмируем его:
lg |
r02 |
= nlg C0A2 |
. |
|
||||
|
|
r01 |
|
|
C0A1 |
|
|
|
Отсюда найдем |
|
|
|
|
|
|
|
|
|
n = |
lg(r01 |
/r02) |
. |
(35) |
|||
|
lg(C0A1 |
|
||||||
|
|
|
/C0A2) |
|
|
|||
Определение порядка реакции по времени полупревращения (частный случай метода Оствальда — Нойеса). Одним из
методов определения порядка реакции по данному веществу является изучение зависимости времени полупревращения от начальной концентрации вещества.
Для реакции n-го порядка (n = 0)время полупревращения t1/2 определяется соотношением
t1/2 |
= |
|
2n−1 −1 |
= |
Q |
, |
(36) |
|||
k |
Cn−1 |
|||||||||
|
|
(n |
− |
1)Cn−1 |
|
|
|
|||
|
|
n |
|
0 |
0 |
|
|
|||
где Q — величина, постоянная для данной реакции. Логарифмируя выражение (36), получим уравнение
lgt1/2 = lgQ−(n−1)lgC0. |
(37) |
Строя зависимость lgt1/2 от lgC0, определяем порядок реакции по угловому коэффициенту прямой (n = 1−tgC0) и константу скорости реакции по отрезку, отсекаемому построенной прямой на оси ординат.
В наиболее простом случае можно оценить порядок реакции по данному веществу, определив время полупревращения для двух начальных концентраций С01 и С02. Тогда, используя уравнение (37), получим
lg t1/2 1 = lgQ−(n−1)lgC01;
lg t1/2 2 = lgQ−(n−1)lgC02.
18
Вычтем из второго уравнения первое:
lg(t1/2)2 −lg(t1/2)1 = (n−1)(lgC01 −lgC02).
После преобразований можно записать:
n = |
lg (t1/2)2 |
/(t1/2)1 |
+1. |
(38) |
|
lg(C01 |
/C02) |
||||
|
|
|
1.5. Влияние температуры на скорость химической реакции
Экспериментальные данные показывают, что скорость большинства химических реакций, как правило, увеличивается с повышением температуры, так как возрастает энергия сталкивающихся частиц и повышается вероятность их химического превращения.
Основное влияние температура оказывает на константу скорости реакции, поэтому, рассматривая влияние температуры на скорость реакции, имеют в виду влияние температуры на константу k. Для гомогенных реакций, протекающих при обычных значениях
температуры (Т < 373 K), в большинстве случаев выполняется эмпирическое правило Вант-Гоффа: при повышении температуры на 10 градусов скорость гомогенной реакции увеличивается в 2 —
4 раза. Отношение констант скоростей, найденных для данной реакции при температурах (Т +10) и Т, называют температурным коэффициентом скорости реакции:
γ = |
kT+10 |
. |
(39) |
|
|||
|
k |
|
|
|
T |
|
|
Если известны скорость реакции при температуре T1 и температурный коэффициент γ, то скорость реакции при температуре T2 можно определить по уравнению
|
|
= r |
γ |
T2−T1 |
(40) |
r |
T2 |
10 . |
|||
|
T1 |
|
|
|
Пользуясь правилом Вант-Гоффа, можно оценить изменение скорости реакции для любого интервала изменения температуры. В среднем для химических реакций γ = 3. Это значит, что при увеличении температуры от 293 до 373 K скорость реакции возрастает в 38, т. е. в 6561 раз.
19
Правило Вант-Гоффа носит весьма приближенный характер.
Гораздо более точным является уравнение Аррениуса, полученное на основе теории активных столкновений:
k = Ze−Ea/(RT). |
(41) |
Согласно теории активных столкновений химическое взаимодействие происходит только при соударении активных молекул, энергия которых Е больше энергии активации (Еa) или равна ей. Экспоненциальный множитель Z в уравнении Аррениуса (см. (41)) можно интерпретировать как долю молекул, энергия которых превышает энергию активации Еa при температуре Т. Уравнение (41) содержит два параметра, не зависящие от температуры: Z — предэкспоненциальный множитель; Еa — энергию активации (R — универсальная газовая постоянная; Т — абсолютная температура).
Молекулярно-кинетическое обоснование теории С. Аррениуса было дано Д.В. Алексеевым, который предложил рассматривать теорию активных соударений, используя закон распределения скоростей Максвелла — Больцмана:
N = N0e−Еа/(RT),
где N — число активных молекул; N0 — общее число молекул. К числу активных молекул относятся молекулы с энергией, большей энергии активации Еa.
Энергия активации (Еa) представляет собой избыток энергии по сравнению со средней энергией молекул при данной температуре, которым должны обладать реагирующие частицы, чтобы вступить в химическую реакцию. Энергия активации измеряется в джоулях (или в килоджоулях) на моль (Дж/моль или кДж/моль).
Множитель Z в уравнении Аррениуса (cм. (41)) согласно теории активных столкновений равен числу столкновений двух молекул за 1 с в 1 cм3 при концентрациях веществ, равных 1 моль/см3, а произведение Ze−Ea/(RT) равно числу активных столкновений, приводящих к химическому взаимодействию. Однако при расчете константы скорости реакции по уравнению (41) получают превышенные значения, особенно для реакций, протекающих в раство-
рах. В связи с этим для объяснения экспериментальных данных в уравнение был введен дополнительный множитель — стерический,
20
или вероятностный, фактор p: |
|
k = pZe−Ea/(RT). |
(42) |
Стерический фактор зависит от природы реакции и может изменяться в пределах 1...10−8. С его помощью учитывают ориентацию молекул в пространстве в момент столкновения относительно друг друга. Стерический фактор p не может быть рассчитан теоретически и для каждой реакции определяется экспериментально. В этом заключается недостаток теории активных столкновений.
Понять физический смысл энергии активации и получить воз-
можность расчета скоростей реакций на основании свойств реагирующих молекул позволяет другая теория — теория абсолютных скоростей химических реакций, или теория переходного состояния.
Основное положение этой теории состоит в том, что при элементарном акте любой химической реакции, например реакции А +ВС → АВ +С, промежуточным состоянием является образование активированного комплекса, в котором в результате сильных соударений происходит перегруппировка валентных связей (одни связи еще окончательно не разорваны, а новые только начали образовываться). Активированный комплекс [АВС]= находится в неустойчивом переходном состоянии и распадается с образованием продуктов реакции:
А +ВС |
|
[АВС]= |
АВ +С |
Cостояние 1 |
Активированный |
→комплекс Cостояние 2 |
Таким образом, химическая реакция представляется как переход системы из состояния 1 (исходные вещества) в состояние 2 (продукты реакции), т. е. как переход через энергетический барьер, равный энергии активации Eа прямой реакции. Обратный переход от продуктов реакции к исходным веществам соответствует еще большему потенциальному барьеру, величина которого равна энергии активации обратной реакции (Eа). Разность энергий активации прямой и обратной реакций представляет собой тепловой эффект процесса: Н = Eа −Eа. Если прямая реакция является экзотермической, то общий запас энергии продуктов реакции меньше, чем запас энергии исходных веществ, т. е. в результате реакции
21