

2 . II начало термодинамики. Энтропия и термодинамическая вероятность
Энтропия – характеризует ту часть полн.энергии системы, кот. не может быть использована для производства работы, представляет собой отработанную энергию.
Согласно второму началу термодинамики энтропия в замкнутой системе постоянно возрастает и в конечном итоге стремится к своему max значению.
Более глубокое толкование понятия энтропии и было дано с позиций статистической физики – если каждое макроскопическое состояние газа может быть получено с определенной вероятностью, то вероятность может быть вычислена через вероятности микросостояний.
Термодинамической вероятностью W называют число микросостояний, которыми может быть осуществлено данное микроскопическое состояние. Свойства термодинамической вероятности похожи на свойства энтропии – обе максимальны в состоянии равновесия, а переход к равновесию связан с их ростом.
Энтропия является придаточной величиной и пропорциональна логарифму термодинамической вероятности: S = klnW – данное выражение определяет принцип Больцмана. Логарифм изменения вероятности совпадает с изменением энтропии. Бесконечное или огромное количество частиц делает невозможным механическое описание и требует описания статистического. Больцман показал, что так как в основе термодинамических процессов лежат обратимые кинетические процессы, то необратимость в термодинамике, измеряемая энтропией, не может быть абсолютной. Таким образом, необратимый процесс есть переход из менее вероятного состояния в более вероятное, а логарифм изменения вероятности состояния с точностью до постоянного множителя совпадает с изменением энтропии состояния.
Второй закон (Второе начало) термодинамики. В изолированных системах самопроизвольно протекают только процессы, не сопровождающиеся уменьшением энтропии (процессы, увеличивающие термодинамическую вероятность).
Математически это записывается так: dSad 0 (Здесь индекс ad означает адиабатический процесс). Равенство нулю возможно при обратимом проведении циклических процессов.
Следует подчеркнуть, что действие Второго Начала термодинамики строго ограничено ТОЛЬКО изолированными системами. А вот понятие энтропии, которое породило этот закон, применимо ко всем видам термодинамических систем и играет важную роль в понимании хода всех термодинамических процессов.
И вне рамок адиабатических процессов энтропия, по физическому смыслу представляющая хаос, если в системе ей не противостоит какая-то противодействующая причина, стремится к возрастанию
Вопрос № 1
Термохимия-раздел химии, изучающий тепловые эффекты различных процессов
З-н Гесса: тепловой эффект (или изменение энтальпии ) реакции зависит только от начальн. и конечн. состояний реагирующих в-в и не зависит от промежуточных стадий реакции.
Теплов. эффект реакции – кол-во выделен. или поглащен. теплоты при протекании реакции
Теплота образования (энтальпия) – теплов. эффект реакции образования одного моля в-ва из прост. в-в, устойчивых при станд. условиях.
Теплота сгорания - это теплота, кот. выделяется при сгорании в-ва в кол-ве 1 моль
Следствия из з-на Гесса:
-Тепловой эффект прямой реакции равен по величине и противоположен по знаку тепловому эффекту обратной реакции (з-н Лавуазье — Лапласа).
- Тепловой эффект хим. реакции равен разности сумм теплот образования продуктов реакции и исходных в-в, умноженных на стехиометрические коэффициенты (ν):
- Тепловой эффект хим.реакции равен разности сумм теплот сгорания исходных в-в и продуктов реакции, умноженных на стехиометрические коэффициенты (ν):
- Если начальное и конечное состояния хим.реакции (реакций) совпадают, то ее (их) тепловой эффект равен нулю.
Применение з-на Гесса к расчету тепловых эффектов: закон Гесса позволяет рассчитывать теплоты образования нестабильных соединений и тепловые эффекты реакции, кот.нельзя осуществить экспериментально . Так,невозможно определить тепловой эффект реакции горения графита до оксида СО, т.к. при этом всегда образуется то или иное количество СО2.
Первое начало термодинамики: если в замкнутой системе,состоящей из неск.тел,имеющих первоначально различн. t, происходит теплообмен, то никакая работа внутри системы не совершается.
Вопрос №3
Термодинамические потенциалы(функции) - характеристическая ф-ция в термодинамике, убыль кот. в равновесных процессах, протекающих при постоянстве значений соответствующих независимых параметров, равна полезной внешней работе.
Энергия Гиббса (изобарно-изотермический потенциал)-свободная энергия системы при пост. давлении и температуре ,обозначают буквой G
Энергия Гельмгольца (или просто свобо́дная эне́ргия) — термодинамич. потенциал, убыль кот. в квазистатическом изотермическом процессе равна работе, совершённой системой над внешними телами.
Второй закон термодинамики дает критерий направленности самопроизвольных необратимых процессов. Всякое изменение состояния системы описывается соответствующим изменением особой ф-ции состояния - энтропии S, кот. определяется суммарной величиной поглощенных системой приведенных теплот Q / T .
В этом и состоит эволюционный критерий направленности необратимых изменений в изолированных системах, кот. всегда идут с увеличением энтропии до ее max значений при окончании процесса и установлении термодинамич. равновесия. Увеличение энтропии означает падение степени упорядоченности и организованности в системе, ее хаотизацию.
Вопрос №17.
Скоростью химической реакции называется количество вещества, вступающего в реакцию или образующегося при реакции за единицу времени в единице объема системы.
Количество вещества выражают в МОЛЯХ, а объем в ЛИТРАХ. В этом случае мы получаем удобную для работы величину - КОНЦЕНТРАЦИЮ вещества в моль/л, которая ИЗМЕНЯЕТСЯ в ходе реакции.
Таким образом, скоростью реакции называют изменение концентрации какого-нибудь вещества, участвующего в реакции, за единицу времени (например, за секунду или за минуту). Отсюда другое определение скорости реакции:
Скоростью химической реакции называется ИЗМЕНЕНИЕ КОНЦЕНТРАЦИИ реагента или продукта в единицу времени.
Разницу между тем, что было и тем, что стало, часто обозначают буквой греческого алфавита D (дельта) Следовательно, только что приведенное определение математически можно выразить так:
![]() ,где
v - скорость реакции, DC - изменение
концентрации (в моль/л), а Dt - интервал
времени, в течение которого это изменение
произошло (сек). "моль/л.сек".
,где
v - скорость реакции, DC - изменение
концентрации (в моль/л), а Dt - интервал
времени, в течение которого это изменение
произошло (сек). "моль/л.сек".
При
этом за ∆c
можно брать изменение концентрации
как одного из реагирующих в-в,так и
одного из продуктов. Но т.к. скорость
реакции,по самому смыслу этого
понятия,должна иметь положит.величину,то
в случае исходных в-в ∆с=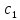 -
-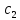 ,где
,где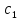 -концентрация
в-ва в начальный момент времени
-концентрация
в-ва в начальный момент времени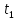 ,
,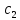 -концентрация
в-ва в конечный момент времени
-концентрация
в-ва в конечный момент времени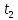 ,а
в случае продуктов ∆с=
,а
в случае продуктов ∆с=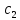 -
-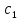 .
На практике под скоростью хим. реакции
обычно понимают именно среднюю скорость.
.
На практике под скоростью хим. реакции
обычно понимают именно среднюю скорость.
Наряду
со средней скоростью пользуются
истинной
скоростью реакции,
под которой понимают производную
концентрации какого-либо участвующего
в реакции в-ва по времени. Напр, для
реакции aA+bB
→dD+eE
скорость в данный момент времени v= , где
, где -мгновенная
концентрация в-ваA.
Индекс T
означает, что процесс совершается при
пост. температуре. Т.к. изменения
концентрации в-в,участвующих в реакции,
связаны стехиометрическим уравнением
реакции, то скорость реакции можно
выразить через изменение во времени
концентрации любого из в-в - как
исходных,так и конечных. Определенные
таким образом скорости связаны между
собой простым соотношением :
-мгновенная
концентрация в-ваA.
Индекс T
означает, что процесс совершается при
пост. температуре. Т.к. изменения
концентрации в-в,участвующих в реакции,
связаны стехиометрическим уравнением
реакции, то скорость реакции можно
выразить через изменение во времени
концентрации любого из в-в - как
исходных,так и конечных. Определенные
таким образом скорости связаны между
собой простым соотношением :
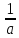 |
|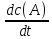 |
=
|
=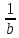 |
|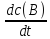 |
=
|
=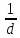 |
|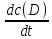 |
=
|
=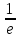 |
|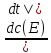
Факторы, влияющие на скорость химических реакций.
1. Природа реагирующих в-в. Большую роль играет х-р хим- связей и строение м-л реагентов. Реакции протекают в направлении разрушения менее прочных связей и образования в-в с более прочными связями. Так, для разрыва связей в м-лах H2 и N2 требуются высокие энергии; такие м-лы мало реакционноспособны. Для разрыва связей в сильнополярных м-лах (HCl, H2O) требуется меньше энергии, и скорость реакции значительно выше. Реакции между ионами в р-рах электролитов протекают практич. мгновенно.
Пример.
Фтор с водородом реагирует со взрывом при комнатной t, бром с водородом взаимод. медленно и при нагревании.
2. Концентрация. С увеличением концентрации (числа частиц в единице объема) чаще происходят столкновения м-л реагирующих в-в - скорость реакции возрастает.
3. Температура. При повышении t на каждые 10°C скорость реакции возрастает в 2-4 раза (Правило Вант-Гоффа). При увеличении температуры от t1 до t2 изменение скорости реакции можно рассчитать по формуле:
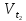 /
/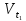 =
=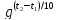
(где
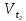 и
и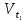 - скорости реакции при температурах
- скорости реакции при температурах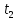 и
и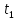 соответственно;g-
температурный коэффициент данной
реакции).
соответственно;g-
температурный коэффициент данной
реакции).
Правило Вант-Гоффа применимо только в узком интервале температур. Более точным является уравнение Аррениуса:
k
= A •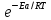
где
A - постоянная, зависящая от природы реагирующих веществ;
R - универсальная газовая постоянная [8,314 Дж/(моль • К) = 0,082 л • атм/(моль • К)];
Ea - энергия активации, т.е. энергия, которой должны обладать сталкивающиеся молекулы, чтобы столкновение привело к химическому превращению.
4. Пов-ть соприкосновения реагирующих в-в. Для гетерогенных систем (когда в-ва находятся в разн. агрегатных состояниях), чем больше пов-ть соприкосновения, тем быстрее протекает реакция. Пов-ть тверд. в-в м.б. увеличена путем их измельчения, а для растворимых веществ - путем их растворения.
5. Катализ. В-ва, кот- участвуют в реакциях и увеличивают ее скорость, оставаясь к концу реакции неизменными, назыв-катализаторами. Механизм действия катализаторов связан с уменьшением энергии активации реакции за счет образования промежуточных соединений. При гомогенном катализе реагенты и катализатор составляют одну фазу (находятся в одном агрегатном состоянии), при гетерогенном катализе - разные фазы (находятся в различных агрегатных состояниях). Резко замедлить протекание нежелательных хим. процессов в ряде случаев можно добавляя в реакционную среду ингибиторы (явл. "отрицательного катализа").
Активные молекулы – м-лы, в кот. энергия равна или больше энергии активации.
Для
того чтобы м-лы могли прореагировать,
они должны обладать запасом энергии,равным
или большим некоторой величины , которая
называется энергией
активации (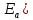 .
.
Вопрос №16.
Раствор – гомогенная (однородная) смесь, образованная не менее чем двумя компонентами, один из кот. назыв. растворителем, а другой – растворимым в-вом, это также система переменного состава, находящаяся в состоянии хим. равновесия.
Растворитель – компонент, агрегатное состояние кот.не изменяется при образовании р-ра. В случае же растворов, образующихся при смешении газа с газом, жидкости с жидкостью, твёрд. в-ва с твёрдым,растворителем считается компонент, кол-во кот. в р-ре преобладает.
Растворенное в-во - компонент жидк. или тверд. р-ра, кот. присутствует в меньшем или незначит. кол-ве.
Р-ры делятся на истинные и коллоидные.
В истинных р-рах (часто называемых просто р-рами) частицы растворенных в-в невидимы ни визуально, ни с помощью оптич. микроскопа. Частицы относит. свободно передвигаются в среде растворителя. В кач-ве растворенных в-в в истин. р-рах могут содержаться неэлектролиты (глюкоза) в виде мол-л и электролиты(хлорид натрия) в виде ионов.
Суспензии – дисперсные системы, в кот. дисперсной фазой явл. тверд. в-во,равномерно распределенное в виде мельчайших частиц в жидкой дисперсионной среде во взвешенном состоянии,причем тверд. в-во практич. нерастворимо в жидкости. Чем мельче частицы,тем дольше сохраняется суспензия. Суспензии относятся к грубодисперсным системам. Пример суспензии – взболтанная глина в воде: со временем частицы выпадут на дно сосуда под действием сил тяжести. В природных условиях образование суспензий происходит при размывании почв и грунтов водой, загрязнении водоемов атмосферной пылью.суспензии широко используют в строительной технологии,в пр-ве керамики,пластмасс,лакокрасочных материалов, бумаги и др. Чтобы приготовить суспензию , надо в-во измельчить до тонкого порошка,высыпать в жидкость , в кот. в-во не растворяется и хорошо взболтать. Чем меньше частички,тем дольше будет сохраняться суспензия.
Эмульсии – дисперсные системы, в которых дисперсные фаза и среда явл. взаимно не смешивающимися жидкостями (иначе – это неоднородная система, состоящая из капель жидкости,распределенных между м-лами др. жидкости). Напр.,из воды и масла можно приготовить эмульсию длительн. встряхиванием. Примером эмульсии также явл. молоко,в кот. мелкие шарики жира распределены в жидкости.
Суспензии и эмульсии – двухфазные системы.
Р-ры подразделяются на:
Молекулярные –водн. р-ры неэлектролитов – органич. в-в(спирт,глюкоза)
Молекулярно-ионные – р-ры слаб. электролитов(азотистой,сероводородн. кис-т)
Ионные – р-ры сильн. электролитов(щелочей,солей,кис-т-NaOH,
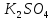 ,HN
,HN )
)
Р-ры бывают концентрирован. и разбавлен.. Из практич. опыта известно, что растворимость многих в-в небезгранична и зависит от t.
Насыщенным называется такой р-р , в кот. при данной t в-во больше не растворяется.
В ненасыщен. р-ре содержится меньше в-ва, а в пересыщен. – больше,чем в насыщенном.
В работе с в-вами важно знать их растворимость в воде. В-во считается хорошо растворимым ,если при комнатной температуре в 100 г растворяется 1 г этого в-ва; если меньше, то в-во считается малорастворимым;при растворимости менее 0,001 г – нерастворимым.
Растворимость – св-во вещ-ва раствориться в воде или др. растворителе.
Хим. теория образования р-ров Д.И.Менделеева, по кот. между растворителем и растворенным вещ-вом происходят хим. взаимодействия (хим. реакции) с образованием особой группы вещ-в: сольватов, гидратов и кристаллогидратов.
В рез-те хим. взаимодействия растворен. вещ-ва с растворителем образуются соединения,кот назыв.сольватами или гидратами, если растворителем явл. вода. Образование таких соединений роднит р-ры с хим. соединениями.
Концентрация р-ра – содержание растворенного вещ-ва в опред. массовом или объемном кол-ве р-ра или растворителя.
Массов.доля растворен.вещ-ва: Ѡ=(m в-ва/m р-ра)*100% m р-ра=ƿ*V (г)
Молярн.концентрация(молярность): СМ=δ/V (моль/л) δ=m/M (моль) CM=m/(M*V) (моль/л)
Норм.концентрация(нормальность): СН= δ/V (экв/л) СН=m/(Э*V) (экв/л) δ=m/Э
Насыщен.р-р – р-р,кот.нах-ся в динамич.равновесии с избытком растворяемого вещ-ва.
Пересыщенный р-р — р-р, содержащий больше растворённого вещ-ва, чем в насыщенном р-ре, избыток вещ-ва легко выпадает в осадок.
Как правило, растворимость тверд.вещ-в в оде с повышением t возрастает. Но имеются и вещ-ва,растворимость кот.при увеличении t понижается. Давление влияет на коэф-т растворимости тверд. тела незначит-но,т.к. при растворении не происходит заметного изменения объема системы. Выделение вещ-ва из раствора при охлаждении – кристаллизация.
При растворении газов в воде выделяется теплота. Поэтому в соответствии с принципом Ле-Шателье при повышении t растворимость газа уменьшается. Растворимость газа от давления определяется з-ном Генри: при пост. t растворимость газа пропорц. его порциональному давлению. Для газов,взаимод. с растворителем,а также при высоких давлениях наблюдаются большие отклонения от з-на Генри.