

§ XI.3. Применение к оценке предэксп в элементарных реанаавк
Рассмотрим применение чета константы скорости ]
(XI.36) для koi [.51) примет вид
dt
' rt2."
(X1.5C
Суммы по состояниям гса G* различаются на Q
354
уравнения
Аррениуса. ляег вычислить стерический
уравнений (XI.20) и (XI.46) с учетом
и
полагая АН*
= Е, получаем
AS?
Xnln2 Kj.
Р= е
LoNA h
(XI.52)
рвческий фактор называют также энтропийным множителем или вероят---=---м фактором [см. уравнение (XI.52)]. Энтропия активации может быть :лена при помощи статистической термодинамики, если известно стро-активированного комплекса. В настоящее время такие расчеты могут выполнены только для простейших реакций, для которых вероятное ie активированного комплекса может быть предсказано теоретически. ; сложных молекул такие предсказания становятся мало надежными, периментально получить сведения о строении активированных комплек-| невозможно, так как это не промежуточное соединение, а только переход-состояние. Отсутствие экспериментальных данных о строении актнвиро-эго комплекса является серьезным недостатком теории абсолютных ско-
реакций и затрудняет ее применение. 1есмотря на ряд внутренних противоречий и отсутствие эксперименталь-данных о строении активированного комплекса, теория абсолютных гей реакций вследствие сравнительной простоты математического ап-является в настоящее время наиболее широко используемой теорией аетики элементарных химических реакций; она позволяет правильно объясняй полуколичественно предсказать много закономерностей для кинети-химических реакций.
имеет вид
§ XI.3. Применение теории абсолютных скоростей реакций к оценке предэкспоненциального множителя в элементарных реакциях
Рассмотрим применение теории абсолютных скоростей реакций для рас-[ щтта. константы скорости некоторых реакций. Для мономолекулярной реакции
А -> А* -»Продукты (XI.53)
Сравнение (XI.36) для константы скорости реакции при Av#=0 и с учетом EQ.51) примет вид
Е
din JS* =d In Я?):
кГ Q* RT
X ~е ■
h Qa
(XI.54)
Суммы по состояниям исходных молекул Qa и активированного Комплекса Q?_ различаются на одну колебательную степень свободы, так как
355
Рис.
XI.5.
Плоскости
сим;
лярные
орбитали
двух

Hg (3Р)+N20 (1Ъ) =» N2 (XZ)+О (3Р)+Hg (XS)
Поскольку в данном случае процесс не ограничен спиновыми и орбитальным* запретами, разложение протекает без осложнений.
К сожалению, класс реакций, подчиняющихся правилам запрета Вигие-ра — Витмера, очень ограничен, хотя многие из них важны, например реакция в пламени, при взрывах и в фотохимии. Попытка распространить эти правила на многоатомные нелинейные молекулы оказалась неудачной.
§ XI.8. Правило сохранения
орбитальной симметрии Вудворда — Хоффмана
и корреляционные диаграммы.
Оценка энергии активации
Существуют реакции, протекающие с высоким потенциальным барьером которые нельзя описать с помощью правил Вигнера — Витмера. Реакция димериации этилена в циклобутан
С2Н4+С2Н4 —» С4Н8
является примером превращения в нелинейной системе, имеющей большут энергию активации, наличие которой не может быть предсказано правилами Вигнера — Витмера.
Вудворд и Хоффман показали, что этот барьер может быть связан с несс-хранением симметрии определенных молекулярных орбиталей в ходе реакции Если занятые орбитали исходных веществ одинаковы по симметрии только с занятыми орбиталями продуктов реакции, то реакция по правилу Вудворда — Хоффмана считается разрешенной и протекает практически без потенциального барьера. Если же какая-либо из занятых орбиталей реагирующих веществ коррелирует с незанятыми орбиталями продуктов реакции (или наоборот), то реакция является запрещенной. При этом она может протекать, но обычно с высоким потенциальным барьером или совсем по другому механизму.
При анализе сохранения признаков симметрии, особенно в сложных случаях, пользуются корреляционными диаграммами. Различают корреляционные диаграммы (КД) орбиталей и корреляционные диаграммы состояний. Корреляционные диаграммы орбиталей — это схема, которая в координатах «энер-
368
![]()
nc. XI .6. Орбитали взаимодевг-
указаны свойства симметрия
гая» — «координата реакгл глеств в орбитали прол; у диаграмме состояний изоб в их изменение в ходе реактива необходимо соблюдать
Правило сохранения симма лей) системы в ходе реакции g и и, + и —).
Правило непересечения, сожги ющие состояниям (орбиталии лри построении КД такое пеан) 134-5861
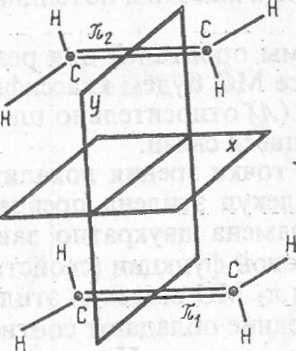
Тип симметрии относительно оси симметрии
Е+ПА х-па
симметрии молекул (см. Вигнера — Витмера их 1>;тся около обозга ш
фотосенсибилизации ртуть [ом состоянии:
[ спиновыми и орбитальньа
правилам запрета Бе: .-е важны, например реакп i распространить эти правя. i неудачной.
потенциальным барьером, за — ВйТмера. Реакции
ie, имеющей большую предсказано правилам*:
может быть связан с несс-биталей в ходе реакция, по симметрии только я по правилу Вудвор-практически без по-танятых орбиталей реагя-продуктов реакция При этом она может барьером или совсем
t особенно в сложных случа-_ Различают корреляционные ■анаграммы состояний. Кор-■оторая в координатах «энер-
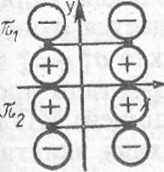
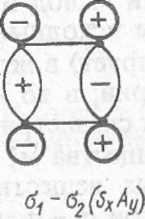
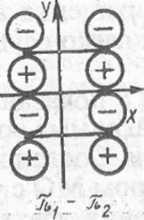
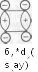
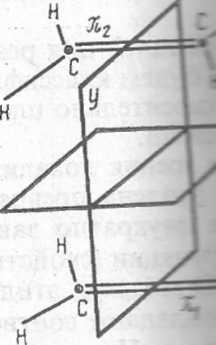
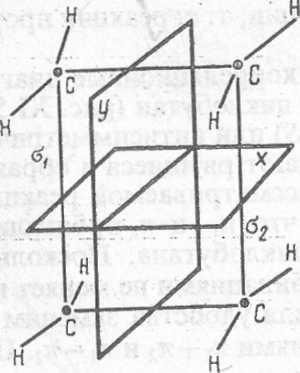
Рис XI.S. Плоскости симметрии, относительно которых классифицируются молекулярные орбитали двух молекул этилена и циклобутана в реакции 2С2Н4-<• С4Н8
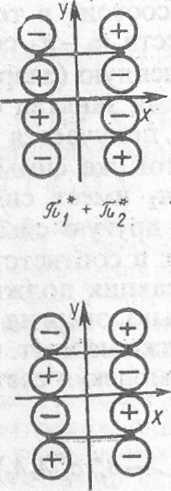
Рис.-XI.6. Орбитали взаимодействующих молекул этилена и циклобутана. В скобках указаны свойства симметрии относительно плоскостей х и у на рис. XI.5
гия» — «координата реакции» показывает переход орбиталей исходных веществ в орбитали продуктов реакции той же симметрии. На корреляционной диаграмме состояний изображаются уровни энергий всей системы в целом и их изменение в ходе реакции. При построении корреляционных диаграмм необходимо соблюдать два правила.
Правило сохранения симметрии волновой функции (или молярных орбита-лей) системы в ходе реакции (спин S, орбитальный момент Я и Л, симметрия g и и, + и -).
Правило непересечения, согласно которому корреляционные линии, отвеча- ющие состояниям (орбиталям) одинаковой симметрии, не пересекаются. Если при построении КД такое пересечение выявляется, то это означает запрет на 24-5861 369
протекание реакции, т. е. реакция протекает с высоким потенциальным ба: = ром.
Рассмотрим-корреляционные диаграммы орбиталей для реакции димет зации этилена в циклобутан (рис. XI.5). Все МО будем классифицировать с симметричные (S) или антисимметричные (А) относительно плоскостей х е которые пересекают рвущиеся и образующиеся связи.
Сущность рассматриваемой реакции с точки зрения локализованных У. состоит в том, что яг и я2-орбитали молекул этилена превращаются в I и о-2-орбитали циклобутана. Поскольку замена двукратно занятых МО з линейными комбинациями не меняет волновой функции (свойство детермнн нта Слейтера), для удобства заменим щ и я2 МО молекул этилена их лиез ными комбинациями 7ii + n2 и щ — пг. Последние обладают соответствующе* свойствами симметрии относительно плоскости х. По этим же причиЕЛ заменим МО ах и а2 циклобутана на их линейные комбинации ai + a2 и ах— t Эти комбинации, а также соответствующие возбужденные МО (с индексом показаны на рис. XI.6; здесь же указана их симметрия относительно плоек стей хиу (Sx, Sy, А„ Ау).
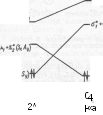
Исходя из схем рис. XI.6, построим корреляционную диаграмму МО (рг XI.7). На ней прямыми линиями соединены МО одинаковой симметрж Главный вывод из нее состоит в том, что занятая (связывающая) орбита: системы исходных веществ щ — п2 симметрии AJSy превращается (говорят коррелирует) в незаполненную (разрыхляющую) МО продукта «rj+<rj той з симметрии; в то же время занятая (связывающая) ах—<т2-орбиталь продух реакции симметрии SxAy получается из незанятой (разрыхляющей) МО исхе ного вещества nf + n* той же симметрии. Как видно, верхняя занятая II исходных веществ пх — п2 имеет симметрию AxSy, а соответствующая II продуктов ffj —о-2 имеет другую симметрию: SxAy, т. е. они не коррелируй между собой. Этот факт в соответствии с правилом Вудворда — Хоффма; указывает на то, что реакция должна протекать с высоким потенциальЕь. барьером. И действительно, энергия активации такого превращения так вел ка, что реакция практически не идет. Однако эта же реакция легко осуществл ется после возбуждения молекул светом.
61-62(sxay) 6f+62(sx5g)
координата реакции
Рис. XI.7. Корреляционная диаграмма молекулярных орбиталей для реакции 2С2Н4- '
>С4НЙ
 На
рис. XI.8
показана корр ляционная диаграмма
состояо той же реакции. Состояние оср
деляется набором МО с указан ем их
заполнения электронам Отличие этой
диаграммы предыдущей состоит в том, ч
вместо уровней МО здесь взял уровни
энергии системы в z
лом.
Симметрия состояе:
определяется
с учетом заполЕ
ния
уровней электронами i
правилу: если два электрона в
ходятся
на двух уровнях с о л
наковой
симметрией S
или
А.
результирующая
симметрч двух электронов 6\z
S(SxS=S,AxA
= S),a.
если
с: на уровнях различной симм; рии, то
SxA=A.
Легко
зам тить, что при
двухэлектронЕ:
На
рис. XI.8
показана корр ляционная диаграмма
состояо той же реакции. Состояние оср
деляется набором МО с указан ем их
заполнения электронам Отличие этой
диаграммы предыдущей состоит в том, ч
вместо уровней МО здесь взял уровни
энергии системы в z
лом.
Симметрия состояе:
определяется
с учетом заполЕ
ния
уровней электронами i
правилу: если два электрона в
ходятся
на двух уровнях с о л
наковой
симметрией S
или
А.
результирующая
симметрч двух электронов 6\z
S(SxS=S,AxA
= S),a.
если
с: на уровнях различной симм; рии, то
SxA=A.
Легко
зам тить, что при
двухэлектронЕ:
370
 dokhm
потенциальным
барье-
dokhm
потенциальным
барье-
жталей для реакции димегг-: л ем классифицировать елг. носнтельно плоскостей х и к, вязи.
зрения локализованных Ml этилена превращаются в г-двукратно занятых МО е яи (свойство детермиЕл-молекул этилена их линей-s обладают соответствующие ~~i х. По этим же причинам ?смбинации ахЛ-аг и ах — с; денные МО (с индексом * рия относительно плоскс-
| диаграмму МО (рис одинаковой симметрии, (связывающая) орбиталъ превращается (говорят — (МО продукта <т*+<т* той та) ах—<г2-орбиталь продукта i разрыхляющей) МО исхол-аатдно, верхняя занятая МО а соответствующая МО т. е. они не коррелируют Вудворда — Хоффмана с высоким потенциальным I превращения так вели-: реакция легко осуществля-
рис. XI.8 показана корре-онная диаграмма состояний се реакции. Состояние опре-[ набором МО с указани-заполнения электронами этой диаграммы от цей состоит в том, что уровней МО здесь взяты энергии системы в це-Симметрия состояния яется с учетом заполне-уровней электронами по если два электрона на-на двух уровнях с оди-: симметрией S или А, то :> тирующая симметрия электронов будет 5 = S, AxA = S), а если они ровнях различной симмет-то Sx.A=A. Легко заме-что при двухэлектронном
СцН8
Рис. XI.8. Корреляционная диаграмма состояний для реакции С2Н4 -* С4Н8
заполнении орбиталей состояние всегда будет S. Поэтому корреляционную диаграмму состояний строят по корреляционной диаграмме орбиталей способом, который виден из сравнения рис. XI.7 и XI.8.
Поясним построение КД состояний по КД орбиталям более подробно. Основное состояние исходного вещества (левый нижний уровень на КД орбиталей) состоит из двух двукратно заполненных МО: (я1+я2)2 и (щ—я2)2. Симметрию этого уровня получаем перемножением знаков симметрии с одинаковыми индексами (х и у): (SxSy)2 (AxSy)2=(Sx х Sx) (Sy x Sy) (Ax x Ax) (Sy x Sy) — =SxSySxSy=(SxxSx)(SyxSy)=SxSy. Такакя же симметрия состояния ф#5у) получится для трех других уровней. Таким образом, из симметрии состояний (рис. XI .8) не видно, какие уровни нужно соединить между собой для получения КД состояний. Для решения этого вопроса нужно об- ратиться к КД орбиталей (см. рис. XI.7). Мысленно переводим электронную пару с МО щ—я2 исходных веществ на МО ffj-t-cj продуктов, а электронную пару с МО а\ — <т2 продуктов — на МО исходных веществ в соответствии с корреляциями (см. рис. XI.7). В результате
получаем возбужденные состояния исходных веществ (щ + я2)2 (я* +яJ)2 (к*—я*) и про-
дуктов (о1+аг)2(а\—<г2)° (<rj + ffj)2 (<xj —а*)0 (пустые МО в последующем опускаем). Из сказанного выше следует, что возбужденное состояние исходных веществ (щ +я2)2 (я* +п%)2 коррелирует с состоянием продуктов {вх+ог}2(с1 — <?г)2, а возбужденное состояние продуктов (в\ + и2)2х x(<7j+o-*)2 коррелирует с состоянием исходных веществ (я1+я2)2(я1 — я2)2. Эти состояния и соединяются на КД состояний прямыми линиями.
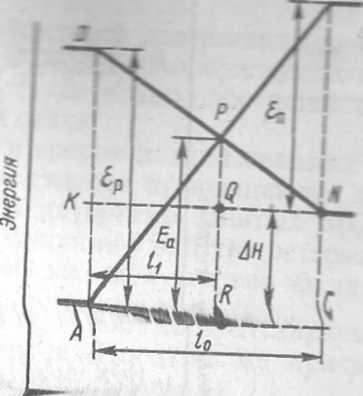
На корреляционной диаграмме состояний энергетические уровни для заполненных и незаполненных МО пересекаются. Положение точки пересечения может быть использовано для приближенной количественной оценке потенциального барьера (энергии активации). Рассмотрим в качестве примера реакцию распада молекулы N20, которая имеет довольно высокий потенциальный барьер, но все же осуществима. Молекула оксида азота (I) сравнительно устойчивое соединение. Разложение на N2 и О начинается только при 1100 — 1200 К. В основном состоянии разрыв связей N — О и N — N при термическом инициировании не может осуществиться из-за запрета по спину (см. § XI.7). На рис. XI.9 приведена энергетическая схема возможного пути протекания реакции
N20(1Z)-.Nj(1Z) + 0(3P)
371
24*

еглеЕня системы Шш сначала сложим их
ШйВл&а значеьгне . энергии активации.
Координата реакции
Рис. XI.10. Схематическое изображение корреляционной диаграммы с пересечением энергетически;, уровней
Переход в продукты может быть осуществлен путем предварительного перевода N20 в другое (возбужденное) энергетическое состояние N20(3n). Прг этом затрачивается энергия Ер и снимается «запрет» по спину на протекание реакции (рис. XT.9, кривая 7)
N20 (1Е) -> N20 (3П) -»N2 (1S)+О (3Р)
Второй путь протекания этой реакции возможен (см. рис. XI.9, кривая 2 когда в результате реакции получается кислород в возбужденном состоянии.
N20 (lZ) -* N2 Czy+OfZ?)
Этот переход разрешен по спину и орбитальной симметрии (см. табл. XI. 1 и XL2). Выделение энергии приводит к переходу продуктов реакции в нор-
Получим выражение для энергии активации Ел через энерге метры исходных веществ и продуктов реакции (рис. XI. 10). т^уголькшшв APR ч& ABC следует
аергии возбуждения исх вестны. Уравнение (Х1.117)«и*и Оценим, например, энержи: элекулы N20. Для этой рои кДж/моль, £п= 193 кДжши 1= 168 к Дж/моль. Энергия ажаи вна 252 кДж/моль, что хороввиц ергии активации. В настоящее время квантовой пользующий корреляпиоБиив зания синтезов органик зных реакций* реакций в [ каталитических прог.