

Глава X
I ФОРМАЛЬНАЯ КИНЕТИКА. СЛОЖНЫЕ РЕАКЦИИ
§ Х.1. Основные понятия
Большинство реакций являются сложными и состоят из нескольких элементарных стадий. При этом все многообразие сложных реакций можно свести к комбинации нескольких типов простейших сложных реакций, а именно двусторонних (обратимых), параллельных и последовательных реакций.
319
Рассмотрение сложных реакций упрощается, если реакция протекает в стационарных или квазистационарных условиях (см. § Х.8) и имеется лимитирующая стадия.
Элементарная (или формально простая) стадия называется лимитирующей, когда закономерности всего процесса определяются в основном кинетическими закономерностями этой стадии. В случае параллельных реакций — это элементарная стадия с константой скоростей, значительно большей коб-стант скоростей для других параллельных стадий. В случае последовательных реакций — это элементарная стадия с константой скорости, намного меньшей констант скоростей для других последовательных стадий. Если в сложное реакции -имеется несколько стадий с близкими по величине константам скорости, то говорят, что в таких реакциях нет лимитирующей стадии. Понятие лимитирующая стадия имеет смысл использовать только для реакций, протекающих в стационарных и квазистационарных условиях.
Математические трудности при установлении кинетических закономерностей сложных реакций значительно уменьшаются, если эти реакции протекаю: В стационарных условиях (для открытых систем) или в квазистационарных условиях для замкнутых систем. Условия протекания сложной реакции в открытой системе называют стационарными, когда процесс протекает в таком режиме, что концентрации промежуточных веществ можно считать постоянными. Это легче и быстрее достигается, когда концентрация промежуточных веществ мала. Условия протекания сложной реакции в закрытой систем; называют квазистационарными, когда в каждый момент протекания такое реакции ее состояние соответствует стационарному состоянию в открытое системе (см. § Х.8).
Задачи, решаемые в формальной кинетике, делятся на прямые и обратные В прямых задачах по известным значениям кинетических параметров сложной реакций^''(константы скорости элементарных или формально простых стадий этой реакции) находят уравнения кинетических кривых, т. е. выражения дл£ зависимости концентрации всех реагентов от времени протекания реакции В обратных задачах, наоборот, по опытным кинетическим кривым находят кинетические параметры (механизм реакции, порядок стадий, значения кое-стант скорости) элементарных или формально простых стадий реакции. . При рассмотрении кинетики сложных реакций делается допущение о независимом протеканий элементарных стадий, т. е. что величина константы скорости элементарной химической реакции не зависит от того, протекают л* в данной системе одновременно другие элементарные реакции. Это допущенкг равносильно предположению, что протекание других реакций не нарушает максвелл-больцмановского равновесного распределения молекул по скоростям или, другими словами, что нарушение равновесного распределения е: скоростям (или по энергиям) между молекулами в системе, из-за химическог: превращения молекул с наиболее высокими энергиями, очень быстро восстанавливается за счет взаимного обмена энергиями при их столкновениях в процессе хаотического теплового движения. Как показывают расчеты и опытные данные, указанное допущение оправдывается в подавляющем чист? реакций, с которыми приходится встречаться на практике. В дальнейшее изложении мы будем полагать, что положение о независимом протеканнг элементарных стадий оправдывается. Заметим, что плазмохимические реакции протекают очень быстро и максвелл •—больцмановское распределеняг молекул при протекании этих реакций нарушается.
Рассмотрим вначале формальную кинетику для сложных гомогенных реакций в закрытьтхсйстемах^^^^^^
Тогда
условие равновесия можно было бы Шт.
для
случая (в)
-ттся на прямые и обратные кских параметров сложной формально простых стадий гривых, т. е. выражения для времени протекания реакция кжнетическим кривым находят ворядок стадий, значения коЕ-иростых стадий реакции. ~: делается допущение о неза-г, что величина константы I зависит от того, протекают rs ie реакции. Это допущенгг других реакций не нарушает мения молекул по скорс-вввювесного распределения г: з системе, из-за химическог: энергиями, очень быстро во: при их столкновение Как показывают расчеты ся в подавляющем числе на практике. В дальнейшее о независимом протекании! что плазмохимические peai-больцмановское распределение ется.
кия сложных гомогенных реи-
§ Х.2. Двусторонние (обратимые) реакции
Двусторонние элементарные реакции первого порядка состоят из прямой и обратной элементарных реакций первого порядка:
*+
А^гВ (Х.1)
где к+ и fc_ — константы скорости соответственно прямой и обратной элементарных стадий. К этим реакциям относятся, например, изомерные превращения в различных классах органических веществ.
Скорость двусторонней реакции (стадии) равна разности скоростей прямой и обратной элементарных реакций:
w=w+— w_=sfe+i, — fc_c2, (Х.2)
где С\ и с2 — концентрация соответственно веществ А и В в момент времени t.
- = k+(ci0-x)-k(c20+x),
Учитывая, что cx = cw—x, с2=с20+х и w=r-^ dcx/dx=dx/dt, получаем
(X.3)
dx
где c10, c20 — концентрации А и В при г=0
(Х.4)
fa ae3aS™ Е~ВаТеЛЬНЫХ ^™ стадий высказанное
1ре^5^к^^^^тПОВе^я ЙК°Р°СТЬ иР^ой элементарной а щто Р РаТН0И' а СК0Р°СТЬ Двусторонней реакции становий
w+~w_ и и>=— =0.
(а)
dt
АлтВ лтС
кроме условия равновесия в виде
щ — w_1 = w2—w_2
(в)
можно представить себе и другие условия, если допустить еще возможность непосредственно превращения С в А, т. е. циклического протекания реакции:
т (б) или
g«
a
записать для случая (б) в виде
W1-W_1 = W2-W_2 = W3-H'_3#0, (Г)
w, = w2=h'3?60 до
ll^0nS^0e теРм°Д™амическое рассмотрение вопроса приводит к вы-ЗУ, что циклическое протекание реакции по механизм? (г)| ™Сд) не может
— 5861
321
![]()
то вещество С является исходным и превращается в вещество А, это прев pair? ние будет происходить по тому же механизму, как и реакция (а), т. е. через п же промежуточное вещество В.
Из уравнения (Х.З) в состоянии равновесия получаем (dx/dt)xmXp = 0;
fc+_c2q + xp fc_ с10-хр'
(X
где хр — изменение концентрации, которое находят из опыта через достаточна большой промежуток времени после начала реакции, когда система достига практически равновесного состояния. Но правая часть уравнения (Х.5) npez-j ставляет собой константу равновесия К, следовательно,
К=
Таким образом, для двустронней элементарной стадии, состоящей из да -щ элементарных реакций, протекающих в прямом и обратном направления константа равновесия равна отношению их констант скоростей.
Из уравнения (Х.З) можно вычислить константы скоростей прямой и с: ратной элементарных реакций двусторонней стадии. Для этого преобразуя его к виду
dx
—= (k+cl0 —к-Сго) — (£+ +к-)х. dt
[бличины L и Я известны из опыта Разделяя переменные в уравнении
dx L-x
\ интегрируя в пределах от 0 до х и «
я=-:
величины L и Н известны из опыта [равнений (Х.6) и (Х.10) находим
Таким образом, обратная зад» Для решения прямой задачи, т квисимости концентраций cj и с2 реакции и константы скорости к.ш 0L11) и (Х.8) в явном виде:
Кещ-
х=-
К—'.
i затем подставляя уравнение (XJ
с\=-
К+1
(х-
Вынося (к+ +к-) за скобки, получим
dt
dx
= (fc++fc_)(L-x),
где
к+сю~ к-С2о
к++к_
Разделив числитель и знаменатель на £_ и учитывая соотношение (X:', приведем выражение для L к виду
с2-
К-1
Значение К находим из cootbm зставляют собой решение пряшия
Если скорость обратной элементграсй р ввательно, составляет небольшую жшш > можно рассматривать с погремиои юстороннюю (например, 100 ■ 0,01 = (Х.1) можно считать практжчеаи Щ
Кс10~с20
К+1
Константу равновесия К находим из опыта:
с20 + хр
К=-
(>
равенства означает, что, пренебр; эту реакцию как одностороннкиа. тш, WOg%. Знак неравенства указывает, чзш: , преобразуем неравенство (Х.16) ж
322
ожение формулируется в веля вновесии в химической реакилй маковой скоростью как си-:: ального равновесия вытекает^ е соотношения концентраций* с, например, если реакцию :
:
1чины L и Я известны из опыта. Разделяя переменные в уравнении (Х.7)
L—x
i интегрируя в пределах от 0 до х и от 0 до t, получаем
*++*._=#,
(Х.10)
в вещество А, это преврати как и реакция (а), т. е. через i
„ 1, ь
Я=- ш = const.
t L-x
(ХМ)
[ получаем (djc/df)*-xp=0;
г личины L и Я известны из опыта и не зависят от времени. Из системы двух 1внений (Х.6) и (Х.10) находим
(X:
НК , Н
к+ =—- и к_ = .
К+1 К+1
(Х.12)
юлят из опыта через достаточн: когда система достигает часть уравнения (Х.5) прел-ательно,
(Хб
й стадии, состоящей из двут и обратном направлениях нт скоростей.
ты скоростей прямой и об-| :ии. Для этого преобразуем
Таким образом, обратная задача решена.
(Х.13)
Для решения прямой задачи, т. е. для получения теоретического выражения висимости концентраций сх й с2 веществ А и В от времени, зная механизм щи и константы скорости к+ и выразим сначала х из уравнений (X. 10), L11) и (Х.8) в явном виде:
x==Kci0-c20 (1_g-(W) K+l
(Х.14)
i затем подставляя уравнение (Х.13) в с1 = Сю—х и с2=С2о+х, получаем
К+1
с10 —с20 , ^10 — с20 -№++*-)'
Ci=——| е
К+1
U-k.)x.
К+1
K(cl0+c20) Kcl0-c20 с2=—— —— е
К+1
(Х.15)
(ХГ
учитывая соотношение (Х.6\
Значение К находим из соотношения (Х.6). Уравнения (Х.14) и (Х.15) едставляют собой решение прямой задачи.
Если скорость обратной элементарной реакции значительно меньше прямой (w_<SCVf+) и, ! следовательно, составляет небольшую долю от нее: w_ —gw+> где, например g=0,01, то такую I можно рассматривать с погрешностью меньше заданной величины 100g% как практичес-[ оддостороншою (например, 100 ■ 0,01 = 1 %). Таким образом, условием того, что двустороннюю акцию (Х.1) можно считать практически односторонней, является неравенство [см. уравнение
ш
gk+ (c10-x)>Ar_(c20+x).
(Х.16)
(X
равенства означает, что, пренебрегая вторым слагаемым в уравнении (Х.З), т. е. рассмат-ая эту реакцию как одностороннюю, мы допускаем погрешность в оценке скорости реакции » в 100g%. Знак неравенства указывает, что эта ошибка меньше 100g%. Учитывая соотношение I (Х.6), преобразуем неравенство (Х.16) к виду
(X
fe+ С20+Х С20 + Х
g—> или g/C>-
k_ сю-х
(X.17) 323
Из неравенства (Х.17) следует, что условиями, которые благоприятствуют тому, чтооиа данную двустороннюю элементарную реакцию или стадию можно было рассматривать еж практически одностороннюю, являются большие значения g, сщ и fc+ или К и малые значения к _ с2о и х.
Очевидно, в начальный период реакции, когда изменение концентрации х (или сте::-превращения-вещества а=х/ею) мало, любая двусторонняя реакция в течение некоторого времеаж может рассматриваться как односторонняя. Максимальное значение хшах, до которого реакпжх можно считать практически односторонней, определим из выражения (Х.17):
xjaax—-
gKcl0-c20 gK+l '
А время fm„, в течение которого реакцию рассматриваем как одностороннюю, находим л выражений (Х.10) и (Х.11), подставляя х^хщдх:
in-
У многих реакций константа равновесия очень велика, т. е. концентрация продуктов реакпг: при завершении реакции во много раз превосходит концентрацию исходных веществ (в равнове; ном состоянии). При этом константа скорости прямой реакции значительно больше констант» j скорости обратной реакции, а кинетика реакции в течение всего времени ее протекания может рассматриваться как практически односторонняя [см. уравнения (Х.16) — (Х.19)]. В общем, нужн; ясно представлять себе, что все элементарные стадии являются в принципе двусторонними; з может быть строго односторонних реакций или стадий. Однако если в рассматриваемых условаг константа скорости обратной реакции в данной стадии сложной реакции во много раз (например на два порядка и более) меньше прямой, то такую стадию считают практически односторонне! и обозначают одной стрелкой, пренебрегая скоростью обратной реакции. Например, если реякттуг | (Х.1) практически односторонняя, то пишут
А-*Вин>=н'+,
При приближении к концу реакции в любой системе, в принципе через бесконечно большое время, должно установиться состояние равновесия. Поэтому при приближении к равновесие, состоянию все стадии сложной реакции становятся двусторонними, а скорости прямых стадий — близкими по значению к скоростям обратных. Одностадийная реакция или лимитирующая стад: -сложной реакции может рассматриваться как односторонняя только в ограниченном интервал изменения концентраций, когда реакция протекает в условиях, достаточно удаленных от cocroi-ния равновесия.
(Х.2: ;
< Следует заметить, что соотношение (Х.6) справедливо только для одностадийной двусторо-ней элементарной реакций. Уже для сложной реакции, состоящей из двух последовательны: | двусторонних элементарных стадий первого порядка
*1 *2
AriPrf В
константа равновесия будет зависеть от четырех констант скорости. Действительно, при pa:- j новесии
.', w=w1-r-h>_i = h'2'~w_2!=0. QL2'.
Выражая скорости прямых и обратных реакций в первой и второй стадиях через концентрации в соответствии с законом действующих масс, получаем в состоянии равновесия
Отсюда
(Х21
kiCA, р-Л^ср, р=0 ик2сР, p-k-гсв, Р=0. ср, р/ср, p=fei/fc_i=^1 или eg, р/сР, р=к2/к.2=к2,
где cj, р -—концентрация i-го компонента в равновесном состоянии; К\ и К2 — константы равно»-! сия для первой и второй стадии. С другой стороны, константа равновесия суммарной реакцо| сА л! св равна К= св. р/с^: р. Учитывая, что (св, р/ср. р) (cPi р/ск р)=eg, р/сд, р, получаем
324
Таким образом, константа пырьмя константами скорости г Л. учебной литературе наиболее — для равновесного состояния гики сложной химической pes t разность прямой и обратной -акают на основе закона i для любой сложной реалии в ■*+сА-£_св; отсюда при i п ми ттт ■тгантыскорости прямой и оС\» шЛй В рассматриваемом случае ■вставить скорость реакции ках центрациям сА и св- Покажем н^^Н ежуточного вещества ста: ■марную скорость образов
(Р) 1
W =и>1+н_;-
гаода cp=(fcicA+fc_2cb)/(fc-i-rt2)-■вой стадии получаем:
Юг это выражение легко
Таким образом, у сложной |овжных реакций в целом; в . I аскньгм образом от констант uupi ' . и jt_ — зависят от концентрация i В литературе есть указания, что i роста двусторонней сложной р— е можно интерпретировать как i иной реакции в состоянии равновесна, I должно получаться термодинамике s концентрации (активности) реагежгэо
§ Х.З. Параллельные реакции
К параллельным pi типам он » для реакции вещество срезе влениях и более. Наггрямеи,
^временно получается о-, м-щ может одновременно протеи
Рассмотрим гомогенную , L одновременно претерпевает я л D, причем обе параллельные я •вый порядок по исходному вея
горые благоприятствуют тому, что! «ню можно было рассматривать е i g, сю и к+ или К и малые значении г
*енение концентрации х (или стез [ реакция в течение некоторого apt.
значение хтш, до которого ред.—-: выражения (Х.17):
(ха
как одностороншою, находку зс
т. е. концентрация продуктов peitza j i исходных веществ (в равн; эе значительно больше констггт всего времени ее протекания меж [ (Х.16) — (Х.19)]. В общем, нуж! ся в принципе двусторонними з Зднако если в рассматриваемых услеьв южной реакции во много раз (напргьг считают практически односторегз зой реакция. Например, если рте га
(Х.23)
Л2*1 ;
Х=——— = «1^2-fc_2fc-i
Таким образом, константа равновесия двустадийной мономолекулярной реакции связана - ттъгрьмя константами скорости прямых и обратных реакций в обеих стадиях.
В учебной литературе наиболее элементарным и наглядным выводом закона действующих для равновесного состояния считается вывод наосновезакона^ де^йствующих мас« для сложной химической реакции при равновесии: w—О. При этом реакцию представляют разность прямой и обратной реакций: w=w+ — tv_, а скорость прямой и обратной реакций т на основе закона действующих масс для химической кинетики, считая, что он примемте любой сложной реакции в целом. Например, для сложной реакции АлгВ находим t+сд—fe-св; отсюда при равновесии сд, р/сд, p=k+/k\.=K, где fc+ и fc-—эффективные
ш скорости прямой и обратной сложной реакции. .;
В рассматриваемом случае двустадийной последовательной сложной реакции (Х.20) Можно вить скорость реакции как разность двух слагаемых, пропорциональных соответственно рациям сд и св. Покажем это. Учитывая, что вблизи состояния равновесия концентрация точного вещества становится очень малой, можно считать процесс квазистационарным, «арную скорость образования вещества Р равной нулю:
... (р) . . . -
' w : -w+w^2~:w-i-*2=kicA+k~2<b—k-icp-k2Cp=Q,
. ci?=№iCa+^-2Cb)/(*-i+*2)- Подставляя это значение ср в выражение, например, для : стадии получаем:
, C-j(*ica+*_2cb) - „ ■
n>=fcieA —— ,v:.'. (Х.24)
еюг это в»^ажёние легко привести к виду и'=^+сд—А:_св, где fc+ =klk2/(kj.1+k2) и к- =k-ik-2/(k-i+k2).
*-i+*2 ;: _ ;
QL2S)
э принципе через бесконечно больавн при приближении к равновес=:чг и, а скорости прямых стадг± I I реакция или лимитирующая < —| только в ограниченном им ции достаточно удаленных от сосавЩ
-:лъко для одностадийной да устам состоящей из двух последователе^
скорости. Действительно, при рг»|
(ХД
второй стадиях через концентрагэ I :остоянии равновесия
(XJ=
«-2СВ, р = 0-
5=^-2 = ^2.
гтеянии; Ki и К2 —константы равн: а
равновесия суммарной реапавт j р)=св, р/сА. р. получаем
Таким образом, у сложной реакции нет констант скорости к+ и к_ прямой и обратной реакций в целом; в действительности это эффективные величины, которые зависят образом от констант скорости всех стадий. Для более сложных случаев коэффициенты Т*-_зависят от концентраций компонентов.
В литературе есть указания, что не всегда имеется возможность представить выражение для рости двусторонней сложной реакции (кинетическое уравнение) в виде двух слагаемых, кото-s можно интерпретировать как скорости прямой и обратной реакций. Но, конечно, для любой вой реакции в состоянии равновесия, когда скорость реакции tv—О, из кинетических уравне-Е должно получаться термодинамическое выражение для константы равновесия через равновес-1 концентрации (активности) реагентов согласно уравнению суммарной реакции.
i Х.З. Параллельные реакции
' К параллельным реакциям относятся химические превращения, когда взя-для реакции вещество претерпевает одновременное изменение в двух равлениях и более. Например, При нитровании бензола
СеД6-г ЖК03=СЛл^02)2+2Н20
ювременно получается о-, м- и й-динитробензол. Разложение тидроксила-может одновременно протекать по двум направлениям:
3NH2OH = NH3 + N2 + 3H20 4NH2OH=2NH3+N20+3H20
Рассмотрим гомогенную реакцию в закрытой системе, когда вещество эдновременно претерпевает мономолекулярное превращение в вещества iD, причем обе параллельные элементарные односторонние реакции имеют вый порядок по исходному веществу А:
325
Решим сначала прямую задачу. Общая скорость образования А равна сумме скоростей реакции по обоим направлениям (с учетом ициил знаков):
dt
= —klcl — k2cl = —(ki+k^Ci,
(X~
где с-\ — концентрация вещества А. Разделяя переменные и интегрируя ура ние (Х.27) в пределах от с10 до сх и от 0 до t, найдем
с\ = с10е
Скорость получения веществ В и D равна
dc2 dc3
—~ = klcl и —=к2сь л
dt dt
где с2 и с3 — концентрации веществ В и D. Подставляя значение сх из в (Х.29), получаем
dc2=fe1c10e"(fcl+*j)'dr и dc3=k2cl0e~<-kl+k2)'dt.
с2--
(1-е
)=
Интегрирование этих уравнений от 0 до с2 (или соответственно от 0 до с-. - ж О до t (предполагая, что в начале реакции с2О=с30 = 0) с учетом (Х.28) дает
(сю—ci);
(c10 —Ci).
(1
) =
fci+fc2
ki+k2
к2сю к{+к2
Выражение для c3 можно получить также из уравнения материального бал (при с20 = с30=0)
С\й~ Сх = с2 +съ.
Отсюда
с3 = с10 — С\ ~С2-
Подставляя в (Х.34) уравнения (Х.28) и (Х.31), получаем уравнение (Х.ЗГ. Найдем селективность реакции (Х.26), разделив уравнение (Х.31) на (XJ
СЗ ^2 С2 + С3 fci+Аг'
где а — интегральная селективность процесса или реакции. Интеграл селективностью процесса при наличии нескольких параллельных реакций на ется отношение концентрации основного (полезного) продукта к сумме кс траций всех продуктов, полученных в результате процесса. Селективв рассматриваемого процесса равна отношению константы скорости оснс реакции к сумме констант скоростей основной и побочной реакций.
326
iEbi из опыта и «** реакции
С ж
Х.4. Последе
— «^iu»i21 u | Я Ж
Г ^ьгражения для кокс ■си последовательно* ои системе с двумя от t стадиями первого
огие сложные ^ гоят из нескольких ■уточные веществе, дующей. В бог-» и однозначно nt ■ —п «тее вероятную схему чет зависимости коаъя f правило, довольно а раций промежуточная эых дифференпналывии едовательными ~
1 другой форме
А -
• — промежуточное соединеаиие, 1 бозначим концентрации веш=етв i w2 — скорости первой и втавввШ ле протекания реакции они могр осмотрим сначала случай, копи* гх стадий близки между собся.
(X.2f
4В
D
А — U
dci dt
Решим сначала прямую задачу. Общая скорость образования веществ, А равна сумме скоростей реакции по обоим направлениям (с учетом принят, знаков):
= - кхсх - к2сх — - (кх + к2)сх,
в ещество
В мы считаем веществом (отходом hi
активность
остается
ещество
В мы считаем веществом (отходом hi
активность
остается
х параллельных pes
Таким образом, прямая -валки веществ А, В и D ащ тлр аллельных реакций извести /Для решения обратной хая Х.35) в
где с, — концентрация вещества А. Разделяя переменные и интегрируя уравв ние (Х.27) в пределах от с,0 до q и от 0 до t, найдем
:-<*•+** (X.:
Ci = ci0e
(X
Скорость получения веществ В и D равна
dc2 , dc3
— = кхсх и —-=/с2сь df dt
М=-
■яестны из опыта и не анизм реакции отвечает
где с2 и с3 — концентрации веществ В и D. Подставляя значение сх из (X.2SJ в (Х.29), получаем
dc2=kxcX0e~(kl^1dt и dc3=k2cXQe~ik^k')1dt. (Х.2
Интегрирование этих уравнений от 0 до с2 (или соответственно от 0 до с3) и rJ О до / (предполагая, что в начале реакции сгй—с30 = 0) с учетом (Х.28) дает
кх+к2
к2с\й кх+к2
C2=ilfli (i_e-fc+*V A- (Cl0-Cl);
кх+к2
(1-е
с3-
-^)=JL^(cxo-cx). кх+к2
Выражение для с3 можно получить также из уравнения материального балг (при с20 = с30=0)
Сщ— сх = с2 + с3. (X
Отсюда
с3 = с10—сх — с2. (X
Подставляя в (Х.34) уравнения (Х.28) и (Х.31), получаем уравнение (Х.32). Найдем селективность реакции (Х.26), разделив уравнение (Х.31) на (X I
——— и с— 02 — ki
Съ к2 С2 + Сз fcl+Zs'
где а — интегральная селективность процесса или реакции. Интегра.'.? селективностью процесса при наличии нескольких параллельных реакций наз? ется отношение концентрации основного (полезного) продукта к сумме кок траций всех продуктов, полученных в результате процесса. СелективЕ: рассматриваемого процесса равна отношению константы скорости основ реакции к сумме констант скоростей основной и побочной реакций, i
326
§ Х.4. Последовательны* виая
Многие сложные pea клин, ci , состоят из нескольких рогата промежуточные вещества, ксггор j в последующей. В большинстзе Ннно и однозначно неизвестен. 1 i наиболее вероятную схему проав Расчет зависимости конце I как правило, довольно сящ центраций промежуточных я "лейных дифференциальЕв=л » ледовательными стадиями 1 схие выражения для концентра юстой последовательной реахя Врытой системе с двумя отвеса стадиями первого порядка.
в другой форме
: Р — промежуточное соединяй Обозначим концентрации: "\ — скорости первой и аз s& протекания реакции сящ; смотрим сначала случай, i " стадий близки между а
скорость образования веществе давлениям (с учетом принять;
1+кг)си
I переменные и интегрируя ура=з^1 .найдем
жЕзество В мы считаем основным (полезным) продуктом, a D — побочным еством (отходом процесса). Как видно, для рассматриваемого процесса Щвективность остается постоянной на протяжении всего времени протекания
ух параллельных реакций.
fci к2
(Х.36)
Таким образом, прямая задача решена. Мы нашли зависимость концент-веществ А, В и D от времени, считая константы скоростей обеих зельных реакций известными. Для решения обратной кинетической задачи перепишем уравнения (Х.28) HX..3S) в виде
ki+k2=M и
М=- In —=const и iV=—=const
t Су Сз
(Х.37)
^■естны из опыта и не должны зависеть от времени, если предполагаемый еханизм реакции отвечает действительности. Из уравнений (Х.36) находим
эдставляя значение Cj из
М , MN
ki= и кг— .
N+1 лг+1
(Х.38)
dt.
ш соответственно от 0 до с3) я . = .-зо = 0) с учетом (Х.28) дает
(сю-ci);
(сю-Ci).
-г
аения материального 6 s
§ Х.4. Последовательные реакции
Многие сложные реакции, с которыми приходится встречаться на практи-состоят из нескольких последовательных элементарных стадий. При этом эмежуточные вещества, которые образуются в одной стадии, расходуются юследующей. В большинстве случаев механизм многостадийной реакции эчно и однозначно неизвестен. Поэтому на основе опытных данных находят *более вероятную схему протекания реакции по стадиям. Расчет зависимости концентрации от времени в последовательных реакци-как правило, довольно сложен, так как для исключения неизвестных дентраций промежуточных веществ обычно приходится решать систему зейных дифференциальных уравнений. Только для более простых реакций эследовательными стадиями первого порядка удается получить аналитике выражения для концентрации веществ в явном виде. Примером такой гой последовательной реакции может служить гомогенная реакция в за-гой системе с двумя односторонними мономолекулярными элементар-стадиями первого порядка
А->Р^В
(Х.39)
получаем уравнение (Х.ЗГ уравнение (Х.31) на Г
в другой форме
А-*Р, Wi=kyCi,
или реакции. Интеграл параллельных реакций нсз. ого) продукта к сумме кт ге процесса. Се лет шва константы скорости осввв и побочной реакций, j
Р-» В, w2 = k2c2, : Р — промежуточное соединение.
Обозначим концентрации веществ А, В, Р в момент времени t через сь с2, с3; и w2 — скорости первой и второй стадии реакции. При нестационарном киме протекания реакции они могут быть различными. Рассмотрим сначала случай, когда константы скорости обеих последова-(ьных стадий близки между собой, например, различаются менее, чем на
327
один-два порядка. Решим прямую задачу, т. е. найдем зависимость конпег раций от времени всех трех веществ, считая известными механизм реака и константы скорости обеих стадий. Скорость образования исходного вешес ва А выразим уравнением
проще это сделать, ищи в стационарном
![]()
(X-
w0) = (— l)Wi или ^-—kiCi.
dt
(X
Его интегрирование (после разделения переменных) от с10 до Ci и от О t приводит к выражению
-к,1
где сю — начальная концентрация вещества А.
Скорость образования промежуточного вещества Р в первой и втор стадиях выражается уравнениями
и43) = (+1)и>ь где щ=кхСи
w(iy^(-l)w2, где w2=k2c2. -
Отсюда суммарная скорость образования вещества Р в обеих последовать ных стадиях равна
ww = Ц3> + >v23>; —=klCl -к2е%. -dt
Подставляя сх из выражения (Х.41), получаем
dt . I
Для решения этого дифференциального уравнения умножим обе его на е*1 и учитывая, что
этом считаем, что в нал
ктавляя в (Х.47) уравноаи
с2 = (
к видно, на кинетиче :тва Р имеется максим щрелеляются из условия с
it2—fc
небольших преобразовав
т=k2/ki. Подставляя (X -: эй
после
ряда преобразо
получим
e — + сък2е =—(c3e 2), dt dt
(X
- (c3e l)=kiClQe 1 . at
Интегрирование уравнения (X.43) от 0 до с2 и от 0 до * приводит к выражен
Разделив обе части на е 2, получим искомое выражение для с3:
с3= (е -е ).
(Х-
Из уравнений (Х.49) ж затах к^ и с3/с10 макова, вещества Р зависит i^H гния констант скорости ста еетической кривой для прсшса 1-^тся к началу координат Zis.
Решение обратной задачи ж ШЛ1) находим
рвагарифмируя
выражение (X
Выражение для концентрации с2 продукта реакции В можно найти дифференциального уравнения скорости образования вещества В:
ЩА
328
•Преобразуя
показатели
нешааа
ж",
;титывая,
что
е^*-х,
а
затем
жшт\
Схй
— С\ = С2
+ Сг.
Врэтом
считаем, что в начале реакции с20=с30=0.
Отсюда
С3-С10-Сх
— С2.
= -кхсх. (Х.-Ч
кяных) от Сю до Сх и от 0 л
ш Н
*:_:ества Р в первой и вторлй]
=кхСи
рства Р в обеих последователе- j zx-k2c3.
b проще это сделать, используя уравнение материального баланса (для акции в стационарном состоянии)
(Х.46)
(Х.47)
(Х.48)
лдставляя в (КА7) уравнения (Х.41) и (КМ), получаем искомое выражение шж с2:
с2=с,о 1-7—г е +——-е ).
Как видно, на кинетической кривой (см. рис. VIII.3) для промежуточного ества Р имеется максимум. Координаты этого максимума (fmax, c3m„) легко (■иределяются из условия dc3/d/=0. Учитывая выражение (КМ), находим
h (-Ье-^+кге^-О. 1
к2-кх
(Х.49)
ie небольших преобразований получаем
In у
*тах—
kx(f-l)
(ХЛ
гния умножим обе его часл
■Р. 7=^2/^1- Подставляя (Х.49) й (КМ), находим концентрацию в максимуме иривой
^К)-< - in у v id у
«10 / у-1 Т^Л
(Х.50)
1Л
Нрн после ряда преобразований*
1-у
I до t приводит к выражен 1).
сражение для с3:
(Х.51)
Из уравнений (Х.49) и (Х.51) следует, что на графике в безразмерных гпординатах kxt и c3jclu максимум на кинетической кривой для промежуточ-г:го вещества Р зависит только от у=к2/кх и не зависит от абсолютного лвчения констант скорости стадий. С ростом отношения к2/кх максимум на ивветической кривой для промежуточного вещества Р становится ниже и сме-гся к началу координат. Таким образом, прямая задача решена. Решение обратной задачи не представляет большого труда. Из уравнения BL41) находим
(X.-
реакции В можно найти ювания вещества В: кх =- In —.
■Г' ^ _ t_ Сх
Логарифмируя выражение (Х.51), получаем
(Х.52)
•Преобразуя
показатели
степени
к
виду
1пу1/(1""у)
и
In''*1"'',
упрощаем
полученное
выраже-i,
учитывая,
что
(^"—х,
а
затем
выносим
уМ1~^
за
скобки.
329
(КАШ
(Х.53
Iny=ln —.
У-1 c3max
Преобразуем уравнение (Х.49) к виду
(Х.54,
fc2=
(Х.55
lny=fc,(y-l)'m«. и, подставляя его (Х.53), найдем значение к2 (при yfc, =&2):
1 . Сю ■■— In .
Если в последовательной реакции (Х.39) &i«&2, то во второй элементарной стадии устанавливается состояние, близкое к равновесному:
как при t=0 в системе приври!
соответствующей равшиви" Кинетическое уравнение дд
«а-1
ie интегрирования (Х.64), у
(Х.56;
Считаем,
что
(Х.57
С2 к2
~= — = &2,
где ЛГ2 — константа равновесия для второй элементарной стадии, т. е. во второй элементарной стадии установилось квазиравновесное состояние. уравнение кинетической кривой для исходного вещества а описывается, очевидно, выражением, подобным (Х.41):
(Х.58
c,=clQe *1' и с10—с, = сш(1—е *''). Из уравнения материального баланса (Х.47) находим
(Х.59)
С2 + Съ = Съ
(1+cl)=Cl0~Cl'
Сонцентрацию с2 вещества В н
c2=(cio+c30)-(ci + c- = эда с учетом (Х.66) получи
Как видно, кинетика ре ах: i константа скорости первой [центраций реагентов А. Р ж В эщей.
(Х.6С
Отсюда с учетом (Х.57) и (Х.58) получаем
сш
с3=——(1-е ) К2+1
(Х.61
и из (Х.57)
■.К2с3=^(1-е-к<).
л2 + 1
Как видно, уравнения кинетических кривых для случая кх < к2 зависят толье в от константы скорости первой стадии, т. е. только первая стадия определяем кинетику реакции (является лимитирующей). кинетические параметры вторе i стадии (к2 и fe_2) в выражения для си с2 и с3 не входят. Видно также, чт; поскольку промежуточное вещество р находится в равновесии с веществом в. к концу реакции при достижении равновесного состояния оно останется в системе.
Теперь рассмотрим случай, когда к2<кх и в первой стадии при протекай in процесса устанавливается состояние, близкое к равновесному:
Следует заметить, что ко для последовательности!
: некоторые последоват енная система дифс s, решена только чис
§ Х.5. Сложные реакции в открытых системах
Сравним степень превращен* акций в аппаратах идеальном реакции, состоящей из двух носа первого порядка
аппарате идеального в и р можно написать, счет
330
Cxi
A Р -> В,
(Х.62)
?ем
(x -
(Х.63)
7*.=*2):
(X-:
как при t=0 в системе присутствует промежуточное вещество в концент-i, соответствующей равновесному состоянию, в первой стадии. Кинетическое уравнение для второй стадии имеет вид
то во второй элемента вновесному:
(Xi
парной стадии, т. е. [овесное состояние. Уг. А. описывается, очевил
(х;
(Х.64)
1 dc3 (-1) at
(Х.65) (Х.66)
ie интегрирования (Х.64), учитывая, что с30=^Г1с10 и сх = съ[Ки получаем
ci=c10e J.
энцентрацию c2 вещества В находим из материального баланса
Clo + Cio = Ci + C2 + C3,
(Х.68)
с2=(с,0 + сзо) - (ci + с3)=с10 (ИГ, +1) - с! (AT, +1)=(Кх +1) (сш - с,). (Х.67) ||гсюда с учетом (Х.66) получаем
с2=(^ + 1)С10(1-е "*).
(Х.5 =
(X
(X.6I
[случая к{<к2 зависят тольег первая стадия определяет аттические параметры втор: -[не входят. Видно также, чт: i равновесии с веществом Е ояния оно останется в сн-
зй стадии при протекай вновесному:
Как видно, кинетика реакции (Х.23) определяется только второй стадией, 1 константа скорости первой стадии кх не входит в выражение для зависимости юнцентраций реагентов А, Р и В от времени. Вторая стадия является лимити-(вующей.
Следует заметить, что аналитические выражения могут быть получены рюлько для последовательности мономолекулярных элементарных стадий, рели некоторые последовательные стадии имеют порядок больше первого, то олученная система дифференциальных уравнений может быть, в общем лгучае, решена только численными методами интегрирования.
§ Х.5. Сложные реакции в открытых системах
Сравним степень превращения и селективность для простейших сложных реакций в аппаратах идеального вытеснения и смешения. В случае сложной [реакции, состоящей из двух последовательных односторонних элементарных стадий первого порядка
к к
А-^Р-^В (Х.69)
в аппарате идеального вытеснения для скорости образования веществ А и Р можно написать, считая для простоты кх — к2=к,
331
(X
—=— key и — = кс,—ксъ, dt dt
где C\ и съ — концентрации_ соответственно А и Р. Интегрирование Ш уравнений от сш и сх от 0 до t дает*:
Концентрацию на вьшя го баланса (Х.73). Для степени превращения и пения (индекс см) получим
-to
График
зависимости : - г
видно,
селективность ■ Ш
сложной
реакции бс: В случае сложной ре
элементарных стадий
(X.-
где С\ж и с3ж — концентрация А и P В реакционной смеси на выходе из "аппарат*] идеального вытеснения; 7 — время пребывания реакционной смеси в аппарате I Концентрацию с2 вещества В на выходе из аппарата найдем из уравнена 1 материального баланса (при с20=с3о=0)
Сгж=с10-сы-сн. (Х.73
Степень превращения вещества А в реакторе идеального вытеснения равна
отношение скоростей сю
«10 —«li cli
авыт = —=1——-=1-
с10 «10
-to
(Х.74)
где индекс «выт» — вытеснение. Считая промежуточное соединение Р за целевой продукт, получаем для селективности в реакторе идеального вытеснения
с1ж
с2ж+с3ж сю-cht
-=kt ——.
I
«выт
(X.75)
Отсюда
*' _. '1 —«выт .
1— «выт
или сгвыт = — 1П
e'-l
П олучим
выражение дл вытеснения остаются форма)
реакции
и для зависимости астеме, если в них
по; 1рате,
т.
е. можно для выходящей из аппарата,
концентраций равно
олучим
выражение дл вытеснения остаются форма)
реакции
и для зависимости астеме, если в них
по; 1рате,
т.
е. можно для выходящей из аппарата,
концентраций равно
и, следовательно, селектнвиосии,
Если реакцию (Х.69) провести в аппарате идеального смешения, то при установившемся (стационарном) Процессе разница между концентрацией вещества, вошедшего и вышедшего из аппарата, как мы видели ранее (см. § IX. 5), равна скорости реакции, умноженной на время пребывания. Поэтому для веществ А и Р можно написать следующие уравнения материального баланса (считая с2о=с30 = 0) [см. уравнение (Х.70)]:
При проведении реаю ного смешения материа В и D можно выразить ур
сю—cix — £7с1ж=0; 0 - с3ж+к! (с1ж - с3ж)= 0.
0 — Cj+tttfA
0 - с-. - '
Отсюда
Отсюда
ci=kllcl;
c3=kjj
А
селективность равна
сю c3ofc7
(Х.76)
l+kt
(1+kt)1
*Для
решения
второго
дифференциального
уравнения
(Х.70)
переносим"
второе
слагаемое
в
левую
часть,
подставляем
q
из
(Х.71)
и
умножаем
обе
части
равенства
на
е*':
dc3
— е™+кс3е*'=кс10
или
d(ciekt)=kciQdt.
dt
Интегрирование
этого
уравнения
дает
(Х.72).
332,
Как
видно, в случае слеша
параллельными
стадиями ztzb
-кс3, (Х.Я
' я Р. Интегрирование эти!
(Х.71
j смеси на выходе из аппарата еахлионной смеси в аппараты вварата найдем из уравнения!
«сального вытеснения равна
(Х.74)]
(Х.75
[■точное соединение Р за деле-1 эре идеального вытеснения
1-W
Концентрацию на выходе из аппарата найдем из уравнения материаль- эго баланса (Х.73). -
И
(Х.77)
Для степени превращения и селективности реакции в аппарате идеального ■иешения (индекс см) получим "
1
l+kl
kt \ + kl
График зависимости а —а для обоих аппаратов представлен на рис. Х.1. раквидно, селективность по промежуточному продукту Р для рассматрива-юй сложной реакции больше в аппарате идеального вытеснения. В случае сложной реакции, состоящей из двух параллельных односторон-элементарных стадий
(Х.78)
Дв Ев
(Х.79)
ргношение скоростей образования продуктов В и D равно
w'l_felcA_fcl
н-2 к2сА к2
Получим выражение для селективности процесса. Для процесса идеального вытеснения остаются формально справедливыми выражения для скорости реакции и для зависимости концентрации реагентов от времени в закрытой зстеме, если в них подставить время пребывания реакционной смеси в аппарате, т. е. можно для концентраций реагентов В и D в реакционной смеси, выходящей из аппарата, использовать уравнения (Х.З5). Отношение этих концентраций равно
cz_ki
(Х.80)
 1льного
смешения,
то при между концентрацией ве-, как мы
видели ранее (см. [ время пребывания.
Поэтому ■ уравнения материального
1льного
смешения,
то при между концентрацией ве-, как мы
видели ранее (см. [ время пребывания.
Поэтому ■ уравнения материального
(Х.76)
С2 __: kV
с2 + съ кх+к2
В, следовательно,селективность
^выт ~
При проведении реакции (Х.78) в аппарате идеального смешения материальный баланс по веществам В и D можно выразить уравнениями (считая с2о=с30 = 0)
0 — с2+1кхсх =0;
0-c3+7A;2Ci=0.
(Х.82)
Отсюда
- - сг ki
C2 = kxtCx\ C-i = k2tC\ и — =r—,
с3 к2
(Х.81)
(Х.70) переносим второе слагаемое ~i равенства на е*:
А селективность равна
с2
кх
с2 + с3 кх+к2
(Х.83)
Как видно, в случае сложной реакции с двумя параллельными стадиями первого порядка селектив-
1 — реактор идеального вытеснения; 2 — реактор идеального смешения
333
ность не зависит от времени пребывания и от типа аппарата. Кроме того, к уравнений (Х.81) и (Х.83) следует, что в обоих аппаратах селективность може: выразить через отношение констант скорости образования двух продуктов.
§ Х.6. Сопряженные реакции
Особую группу представляют собой сопряженные реакции, когда самопрс-1 извольно идущая в системе реакция вызывает протекание другой реакции, ле осуществимой в отсутствие первой (Н. А. Шилов). Это явление называете! химической индукцией. Поэтому две реакции, одна из которых индуктирует протекание другой, называются сопряженными.
Сопряженную реакцию можно представить в виде схемы
А+В -»М; А+'С -+ N; А+В+С -»M+N
Вещество А, реагируя с веществом В, дает вещество М. Вещество А в отсутствие вещества В не взаимодействует с веществами С, но при взаимодействгг веществ А, В и С образуются вещества М и N. Таким образом, вещество Б реагируя с веществом А, вызывает реакцию между веществами А и С. Вещество А, участвующее в обеих реакциях, называется актором; вещество В, реагирующее с актором и индуцирующее реакцию А с С, называется индуктором. Вещество С, взаимодействие которого в системе с актором возможнг только при наличии химической индукции, называется акцептором.
Количественная характеристика эффективности химической индукции, е~ I зываемая фактором индукции Ф, равна отношению скорости расходовангх|
ф=
(X.&5J
акцептора V** к скорости расходования индуктора и>(и):
= W(VH).
Сопряженные реакции осуществляются в том случае, если промежуточны вещества первой стадии служат исходными для последующей стадии, вступи во взаимодействие с акцептором.
HBr02+H2S04 •HBrO + H2S04 ♦ HBr+H2S04
Примером сопряженной реакции может служить реакция между бромновг той кислотой и смесью сернистой и мышьяковистой кислот. Кислота НВгС непосредственно окисляет H2S03, но не окисляет HjAsOj. При действии НВгС на смесь H2S03 и НзАвОз окисляются обе последние кислоты. Это становита понятным, если считать, что процесс окисления H2S03 идет по стадиям:
HBr03 + H2S03-HBr02 + H2S03-HBrO+H2S03
Промежуточные вещества НВЮ2 и НВгО окисляют мышьяковистую кислот? до мышьяковой кислоты. В рассматриваемой реакции НВг03—актор, H2S03 — индуктор и HjAs03 — акцептор. Таким образом, промежуточное активное вещество связывает первичную и вторичную реакции и обеспечивает их совместное протекание. Функции актора, индуктора и акцептора не являются свойствами, закрепленными за теми или иными химическими соединениями. Одно и то же вещество, в зависимости от партнеров, может играть ран актора, индуктора или акцептора. В реакции, где смесь H3As03 и MnSC. окисляется перманганатом калия, Н^вОэ — индуктор, а при взаимодейств^е H3ASO3 со смесью Н2Сг04 и НВгО мышьяковистая кислота — актор.
В сопряженных химических самопроизвольном процессе, им образования вещества в реахпии| I т. е. термодинамически невоэния
§ Х.7. Автокатапитические
Реакция называется авттмт шродуктов реакции. При автои I реакции) не остается постоим (один из основных признаков жа ра, ее неизменность к коь строго говоря, автокатализ не < [ваем его в этом разделе.
Примером автокатализа вк водной среде:
СН3СООС2Н5+Н»
где
Н+
— продукт реакции,
ко Таким образом, эта реакции я
дукт-катализатор.
Значение автокаталю J возросло с разработкой не 1). Поэтому мы остановами , Зависимость скорости ад мени и других факторов, а ■различный вид для разных стовий ее проведения: в завив! закрытых систем является гюм ■вакцин от концентрации каталя [периода, если начальная Рассмотрим автоката: системе и состоит из двух стадвн
I I *.
' A+B-+D-
D -»2Б -А-*В
р А — исходное вещество; D 1лизатор. Первая элемаввя дая.
Выражение для скорости 3 иродукта-катализатора В
■не с2 — концентрация продукта ■Ввцентрация сх исходного зега Шегко видеть, что
334
типа аппарата. Кроме того, i _ аратах селективность може [ образования двух продуктов.
В сопряженных химических реакциях энергия Гиббса, выделяемая при самопроизвольном процессе, в котором участвует актор, расходуется для образования вещества в реакции, протекающей с увеличением энергии Гиббса, т. е. термодинамически невозможной при отсутствии сопряженной реакции.
je реакции, когда самопр:-нротекание другой реакции, иг ~эв). Это явление называется одна из которых индуктирует
в виде схемы
■C-»M+N (Х.84)
1М. Вещество А в отсутст-С, но при взаимодействия Таким образом, вещество В веществами А и С. Вещест-актором; вещество В, ре-А с С, называется индук- \ ^системе с актором возможно ся акцептором. химической индукции, на-скорости расходования!
[ последующей стадии, вступая
• реакция между бромвоэа-гой кислот. Кислота НВгО ■ HjAs03. При действии НВгО; едние кислоты. Это становится H2S03 идет по стадиям:
r+H2S04 -H2S04 hH2S04
дяют мышьяковистую кислоту ■ой реакции НВг03 — актор, образом, промежуточное | реакции и обеспечивает тора и акцептора не являют-химическими соединени-партнеров, может играть роль где смесь H3As03 и MnSO, ктор, а при взаимодействии : кислота — актор.
§ Х.7. Автокаталитические реакции
Реакция называется автокаталитической, если она ускоряется одним из продуктов реакции. При автокатализе концентрация катализатора (продукта реакции) не остается постоянной — она увеличивается, т. е. не выполняется один из основных признаков катализа постоянство концентрации катализатора, ее неизменность к концу каталитического акта (см. § XV.1). Поэтому, строго говоря, автокатализ не относится к разделу «катализ» и мы рассматриваем его в этом разделе.
Примером автокатализа является омыление сложного эфира в слабокислой водной среде:
СН3СООС2Н5+Н2+Н+ сн3соо- +С2Н5ОН+2Н+
где Н+ — продукт реакции, который участвует в реакции со сложным эфиром. Таким образом, эта реакция является автокаталитической, а ион Н+ —продукт-катализатор.
Значение автокаталитических реакций за последние 10 — 15 лет существен- но возросло с разработкой учения о самоорганизации материи (см. приложе- ние 1 ^Поэтому мы остап реакций немного более подроб-
3 аКрЫТЫХ СИСТеМ ЯВЛЯеТСЯ гшяилсйис " "маКСйлсГу Mix "па " ЗаЬхЛ«£*1&^1^* J.
реакции от концентрации катализатора или времени и наличие индукционного периода, если начальная концентрация продукта-катализатора мала.
Рассмотрим автокаталитическую реакцию, которая протекает в з&крытвк системе и состоит из двух стадий:
А+В -»D — лимитирующая стадия;
(а)
D -> 2В — быстрая стадия; А -»В — суммарная реакция,
где А — исходное вещество; D — промежуточное соединение; В — продукт-катализатор. Первая элементарная стадия — односторонняя и лимитирующая.
(Х.86)
Выражение для скорости реакции, а также для скорости образования продукта-катализатора В имеет вид:
w=dcijdt=&ic2 (с0 — с2),
где с2 — концентрация продукта-катализатора В в момент времени /; с0—с2 — концентрация cj исходного вещества А в момент времени t. Покажем это. Легко видеть, что
c0=Cio+C2o=Ci + c2, (Х.87)
335
где
c0—сумма
начальных концентраций (сю+сд,) веществ
А и В. Велича
с0
представляет собой так называемый
инвариант реакции, так как сум} пивые
веществ А ■
концентраций
веществ А и Б в любой момент времени
постоянна. Ден Кинетические KP~^L
крИвые
имеютi
вительно,
из суммарной реакции (а) видно, что при
уменьшении концентрап Д°,
этикинетическ Такие
кря
вещества
А на величину х
на
такую же величину увеличится концентрат,
юсительно средней. (Х91)
ле—
продукта-катализатора В. Очевидно,
что сумма их концентраций в люб Из
Уравнения логи^^^ ^
момент времени
будет оставаться постоянной.
Отсюда юстроить гра*^
шнение
(Х.91):
Из
выражения (Х.86) следует, что график в
координатах w-c2
имеет куполообразной кривой (рис. Х.2,
а) с максимумом (wm„,
е^), нача скоростью, равной wv=fciC20Cio
(при
/=0) и конечной — wx=0
(при7-»со, с2=с0).
Действительно, исследование функции
и>(с2)
на наличие мака (dw/dc2
= 0) дает координаты максимума на графике
(рис. Х.2, а):
С2т==со/2
и
w^fc/Vei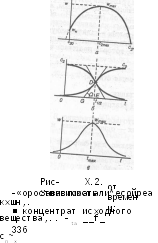 Получим
далее
кинетические
уравнения
для
компонентов
А
и
В
реакции.
Из
уравнения
pi
находим
после
разделения
переменных
Получим
далее
кинетические
уравнения
для
компонентов
А
и
В
реакции.
Из
уравнения
pi
находим
после
разделения
переменных
fc,d<=dc2flc2(c0-c2)].
Легко видеть, что
Vfo(to-C2)}= =[11с2+1Кс0-с2)Уса.
Поэтому уравнение (после ра: ления переменных) преобразуй к виду
cofctd<=dc2/c2 +dc2/(c0—сг).
Проинтегрировав это уравнение, лучаем:
coifcit=In (с2/с20) - In (с0 - c2/ci оУ
Тогда выражение для с2 (/), учитьп что сю=со— сю, будет иметь вид
сг-со/Ксю/сго)* +4-
Величину с2=Со/2 в момент вре> <1/2, определяем из соотношения
сю/сго=exp (cofci lip)-Следовательно, выражение кв ческой кривой для продукта-ка затора В имеет вид:
где
C2sCo/(z+l),
ta*=-W('-rI/2).
рафик, построенный по уравнению < ido, график в координатах w — t ныв я производная логисты. Максима.-.: 1= 1) описывается выражением Щ по ожидать, с выражением (Х.89).
скорость реакции достигается цества становится равной с0/2. Из
![]()
ри /-» + со имеем z-*0 и w-»0. Если концентрация продукта-катам отекания рассматриваемой автот ачале практически не протекает. Ъукцшг, он характерен для автокат
количественная характеристика о шитического метода определения Щ укции отождествляют с моментам:j : реакции, когда метод анализа псам юнной смеси.
Пример Х.1. Провести исследование кжкш ализатора: а) показать, что константа я rose ( касательной в точке перегиба логист^ н > уравнение логисты в безразмерном шв внения логисты универсально (например г -вейной анаморфозы логисты и по ясен—i i лнению определить значения постоянных а Решение, а) Для треугольника GDE щ сматриваемом случае I>E—cq/2. Обозам
уравнение (Х.89)]. Отсюда fcicЈ/4=: б) Перепишем уравнение логисты в зшж
, что сф\ =25, получаем вместе утв
означим 2с2/со=Д и (t—hft)/S = oi. Tcrzi я
■5861
где
с0—сумма
начальных концентраций (сю+см)
веществ А и В. Величин-Со
представляет
собой так называемый инвариант реакции,
так как су концентраций веществ А и В
в любой момент времени постоянна,
вительно, из суммарной реакции (а) видно,
что при уменьшении концентрацим вещества
А на величину х
на
такую же величину увеличится концентраши
продукта-катализатора В. Очевидно, что
сумма их концентраций в любе* момент
времени будет оставаться постоянной.
Отсюда
Јj=Co—сг. (Х.8!
Из выражения (Х.86) следует, что график в координатах w—c2 имеет be: куполообразной кривой (рис. Х.2, а) с максимумом (wmax, с2т), начальное скоростью, равной w„=kiC2oCi0 (при t—О) и конечной — wr=0 (при т-+сс, е1=й с2=с0). Действительно, исследование функции w(c2) на наличие макеимумь (dw/dc2 = 0) дает координаты максимума на графике (рис. Х.2, а):
Сьп = Со/2 и ^,^=(^1/4)^0.
(Х.89
Получим далее кинетические уравнения для компонентов А и В реакции. Из уравнения (Х.И находим после разделения переменных
kidt=dc2/[c2(c0-c2)].
 Легко
видеть,
что
Легко
видеть,
что
Щс2(с0-с2)] = =[1/с2+1/(со-с2)]/со. (Х.9С
Поэтому уравнение (после раазг-ления переменных) преобразует:. к виду
cakidt=dc2/c2 +dc2/(c0-cj).
Проинтегрировав это уравнение, п:-лучаем:
cofet<=1п (с2/с20) -In (с0-с2/с10).
Тогда выражение для с2 (t), учитывах. что сю=со—его, будет иметь вид:
сг-соЖсю/сзоУ 1 -И].
Величину с2=Со/2 в момент времен! Г]/2, определяем из соотношения
cio/c2o=exp(c0fei<i/2).
РС9:
Следовательно, выражение кинет: ческой кривой для продукта-ката.тг затора В имеет вид:
с2=со/(г+1),
где
б — концентрации исходного вещества; в—In —
lnz=-c0fc,/(<-<i/2). (Х-5:
Из соотношения (Х.88) получаем выражение для кинетической кривой исходного вещества А:
веществ А и В. Величин* I реакции, так как сумм* | времени постоянна. Дейет-1 рш уменьшении концентрации у увеличится концентрации j их концентраций в любе* >да ■-■ :. j
(Х.8;
эрдинатах w—с2 имеет вил
юм (и>ти, с2т), начальной
wr=0 (прит~*0о, ci = 0,j
с:) на наличие максимума (рис. Х.2, а):
(Х.89)
i \ в В реакции. Из уравнения (ХМ
Легко видеть, что
1/[с2(со-с2)]= =[1/с2+1/(с0-с2)]/со. (X.*:
эму уравнение (после разде-переменных) преобразуется ; яду
cokidt=dc2/c2 +йсг/(с0-cj). егрировав это уравнение, пс-
с£х t=In (с2/с20) - In (с0 - с2/сю).
. выражение для с2 (/), учитывав | сю=с0—си, будет иметь вид:
—eje t
■ в2-с0/[(Сю/С2о)« '+!]•
с2=Со/2 в момент временя определяем из соотношения
сю/с2о=exp (cofy <i/2).
где
Следовательно, выражение кинетической кривой для продукта-катализатора В имеет вид:
c2=c0/(z+l), (Х.91
mz=-c0A:i/(<-<i/2). (Х.92;
Из соотношения (Х.88) получаем выражение для кинетической кривой исходного вещества А:
X
53
с,=со/(г-Ч1).
I Кинетические кривые веществ А и В представлены на рис. Х2, о. Как дадно, эти кинетические кривые имеют вид S-образных кривых, симметричных гтносительно средней точки. Такие кривые называют «логистой».
Из уравнения логисты (Х.91) легко получить выражение для скорости ■ построить график зависимости скорости от времени w(f), дифференцируя Сравнение (Х.91):
■■кхС&Кг+Л)2. (Х.94)
Трафик, построенный по уравнению (Х.94), представлен на рис. Х2, е. Как видно, график в координатах w —- t имеет вид симметричного пика и называ- ется производная логисты. Максимальное значение скорости реакции при f=f1/2 ш z— 1) описывается выражением Которое совпадает, как и следо-
(Х.95)
вало ожидать, с выражением (Х.89). Как видно из рис. Х.2, а и б, максимальная скорость реакции достигается при ^=^/2» т. е. когда концентрация вещества становится равной с0/2. Из полученных выше соотношений находим
^it=(llcoki)]n(c2olci0).
При t-* + со имеем z-*0 и w->0.
Если концентрация продукта-катализатора В в начальный период времени протекания рассматриваемой автокаталитической реакции мала, то реакция вначале практически не протекает. Этот период времени называют периодом индукции; он характерен для автокаталитических реакций в закрытой системе. Его количественная характеристика обычно связывается с чувствительностью аналитического метода определения продукта-катализатора. Конец периода индукции отождествляют с моментом времени протекания автокаталитической реакции, когда метод анализа позволяет обнаружить его появление в реакционной смеси.
Пример Х.1. Провести исследование кинетической кривой [см. уравнение (Х.91)] продукта-катализатора: а) показать, что константа скорости реакции fcj связана с отрезком 5 (отрезком на оси t касательной в точке перегиба логисты при с2 = с0/2) простым соотношением 5coki=>2; б) получить уравнение логисты в безразмерном виде; в) показать, что отношение сг/со в любой точке уравнения логисты универсально (например, в точках А я В на рис. Х.2, б); г) получить уравнение линейной анаморфозы логисты и пояснить, как на основе „графика,, построенного по этому уравнению определить значения Постоянных коэффициентов k\, 1щ. ;
Решение, а) Для треугольника GDE (см. рис. Х.2, б) имеем соотношение DEjGE=Xg в. В рассматриваемом случае DE=c0/2. Обозначим отрезок GE через 6. Тогда:
<«0-(dc2/df)/.,1/j-h'-^ft:15-2
(Х.96)
[см. уравнение (Х.89)]. Отсюда fc[c^/4 = co25 или
Cofc]5=2.
б) Перепишем уравнение логисты в виде
сг/со=1 4-1).
Учитывая, что cfc\ =26, получаем вместо уравнения (Х.92)
lnz=2(r-/i/2)/5.
(Х97) 337
Обозначим 2c2/c0=/J и (<—*i/2)/<5=a. Тогда уравнение лОгисты примет вид:
2
ехр(-2а)+1
22-5861
где а— безразмерная величина, выраженная в единицах отрезка 8 (при этом за начало отсчг времени принят момент времени tip); Р концентрация с2, выраженная в долях от со/2 (полоз высоты логисты). Поэтому при f = fi/2 к=0 и /?= 1.
в) В точках А и В логисты (см. рис. Х.2, б) значения /?, рассчитанные по формуле (Х.9Т) ~ш «1 = 1 и й2=2, равны соответственно /?]=0,238 и &=0,036. Значит, безразмерные концентраМЩ с2 в точках А и В логисты в два раза меньше и равны соответственно 0,119 и 0,018 доли от с-
г) Преобразуем уравнение логисты (Х.91) к виду:
In[(c0-c2)/c2]=lnz.
Обозначив
лествляется в цепных ви этих реакций в открытых i (приравнивается нулю алгебражтя | вания) каждого промежуточного! кях уравнений, из которых сова [а скорость реакции можно вьфии В качестве примера расонов] | двух односторонних элементарва
ы=(1/со)ш[(с0-с2)/с2],
получим искомое уравнение линейной анаморфозы логисты:
и=*1*1/2-Ы
График этого уравнения в координатах u—t имеет вид прямой линии под тупым углом к оз абсцисс. Угловой коэффициент этой прямой равен tg0= — кх, а отрезок на оси ординат при t—l равен fci<i/2- Отсюда находим fcj и fy2.
Известны также гетерогенные автокаталитические реакции, катализируемых конечными продуктами. Например, растворение меди и ртути в азотнсЁ кислоте катализируется ионами NOJ и оксидами азота; восстановление некоторых твердых оксидов металлов водородом или оксидами азота катализируется образующимся металлом; взаимодействие твердого оксида серебра с СС: катализируется образующимся Ag2C03.
Пусть реакция протекает грация исходного вещее <системы этим веществом тгтвпг 1 в системе установится сташвмвв вещества Р достигнет постояв ■РС.99) получим (cA=cAo=caaBtlj
dep d*
Выясним, через какое i ный режим. В начальный : ного вещества В определяем
§ Х.8. Стационарное и квазистационарное протекание реакций
Для кинетического описания даже простейшей двустадийной односторонней мономолекулярной реакции, протекающей в закрытой системе, приходится составлять и решать систему дифференциальных уравнений. Если число стадий превышает две и некоторые из них являются двусторонними или бимолекулярными и тримолекулярными и особенно для гетерогенных реакций, математические выражения усложняются и решение нельзя получить в аналитическом виде. Однако для определенных типов реакций и в определенных условиях процесс может протекать в стационарном или квазистационар-ном режиме и тогда кинетические расчеты существенно упрощаются.
Стационарный режим протекания реакции может установиться в открытой системе. При этом концентрации всех промежуточных веществ в данной точке пространства остаются постоянными. А это означает, что скорости образования и расходования всех промежуточных веществ одинаковы в течение всего времени протекания процесса в стационарном режиме (в данном сечении в реакторе идеального вытеснения и в любом месте аппарата для реактора идеального смешения):
Проинтегрируем уравнение С что при г=0 сро=0
В результате интегрировав!
In-
отсюда
dc,-
с,=const; — = 0,
(Х.98)
Разделив уравнение (ХЛ*
где с, — концентрация i-го промежуточного соединения в момент времени t.
Для того чтобы стационарный режим протекания реакции установился достаточно быстро и затем сохранился, промежуточные вещества должны быть достаточно активными, т. е. продолжительность их жизни должна быть небольшой, по сравнению с временем протекания реакции. Их концентрация при этом будет достаточно мала. Стационарный режим на практике часто
338
Как видно, формально л достигается только через оеш
отрезка <5 (при этом за начало отсчга , выраженная в долях от со/2 (полозеш
f, рассчитанные по формуле (Х.97) пж Значит, безразмерные концентром --пенно 0,119 и 0,018 доли от с;.
^юществляется в цепных реакциях и при гетерогенном катализе при протекании этих реакций в открытых системах. При этом согласно уравнению (Х.98) приравнивается нулю алгебраическая сумма скоростей получения (и расходования) каждого промежуточного вещества и получается система алгебраических уравнений, из которых концентрации промежуточных частиц, а затем ж скорость реакции можно выразить через концентрации исходных веществ.
В качестве примера рассмотрим двустадийную реакцию, состоящую из двух односторонних элементарных стадий первого порядка:
А-»Р-»В
(Х.99)
[ прямой линии под тупым углом к ос -кь а отрезок на оси ординат при ;=
ае реакции, катализируе-ае меди и ртути в азотнс* : азота; восстановление неко-оксидами азота катализир-. -! твердого оксида серебра с СС-
(X.100)
Пусть реакция протекает в реакторе идеального смешения, причем концентрация исходного вещества А поддерживается постоянной путем подпитки системы этим веществом извне. Через некоторое время после начала реакции в системе установится стационарный режим и концентрация промежуточного вещества Р достигнет постоянного значения сР(сг). Для уравнения реакции (Х.99) получим (сА=сАо=const)
dcp fci
——=К\сдо fc2cP(cr)=0; cP(cr)=— сА0=const.
at «2
Выясним, через какое время после начала реакции установится стационарный режим. В начальный период реакции скорость образования промежуточного вещества В определяем из выражения
двустадийной односторсн-закрытой системе, приходжт-ьных уравнений. Если чес -^являются двусторонними иди —[о для гетерогенных peaks ■ решение нельзя получит* кх типов реакций и в определее-Щтвнарном или квазистациона:-тно упрощаются, установиться в открыт: ■очных веществ в данной точс ier, что скорости образовг-одинаковы в течение всегс и режиме (в данном сеченгж месте аппарата для реактора.
Вившения в момент времени ; ттекания реакции установился ■ежуточные вещества должеъ: ■ьность их жизни должна быть [ реакции. Их концентрации I режим на практике чаете
dcp
It
(X.I01)
(X.102) (X.103)
(X.104)
Проинтегрируем уравнение (Х.101) в пределах от 0 до / и от 0 до сР, приняв, что при г=0 сро=0
In
^lcao
В результате интегрирования находим kiCM-k2cp
\ «1СА0/
отсюда
cp=cao--(1-е ').
*2
Разделив уравнение (X.103) на (X.100), получим
Как видно, формально стационарный режим, т. е. равенство Ср=сР(„), достигается только через бесконечно большой промежуток времени:
22*
339
Однако если мы зададим достаточно малые, но конечные отклонения сР от Ср(СТ)> то они окажутся достижимыми за вполне приемлемое время. Условимся например, считать близкой к стационарной концентрацию Ср^О^Зср^). Тогдг из уравнения (Х.104) находим
; сд.— концентрация Подставляя значенн:
«2 \ сР(ст)у кг
(Х.10:
Нпзода
Например, для кг—103 с"1 получаем t0i9<~3 ■ Ю-3 с, т. е. время достижение стационарного состояния оказывается малым.
Метод расчета с использованием условия (Х.98) при постоянстве концентрации исходных веществ называется методом стационарных концентрации Боденштейна.
При протекании многостадийной реакции в закрытой системе концентрации промежуточных веществ не могут оставаться постоянными, так как концентрации исходных веществ уменьшаются со временем. Однако для реакций в закрытых системах оказывается полезным понятие квазнстационарногс режима реакции. Это режим, при котором концентрации промежуточных веществ в ходе процесса в каждый момент времени отвечают условиям стационарности по отношению к изменяющимся концентрациям исходных веществ, т. е. отношение концентраций промежуточных и исходных вещесп остается постоянным. Поэтому условие (Х.98) применимо и для квазистационарного режима протекания реакции.
Очевидно, что для осуществления квазистационарного режима требование к высокой активности промежуточных веществ, непродолжительности их жизни и низкой концентрации имеет еще большее значение, чем при стационарном режиме. Только при этом условии концентрации промежуточных частиц смогут «подстраиваться» к меняющейся концентрации исходного вещества в каждый момент времени и принимать значения, требуемые при стационарно к протекании процесса.
Оценим время достижения квазистационарного состояния для модельное реакции (Х.99) при условии протекания ее в закрытой системе, т. е. когдг. концентрация исходного вещества уменьшается со временем. Кинетику этсг реакции см. в § Х.З. Концентрация промежуточного вещества меняется сс временем и процесс в течение этого периода реакции (до максимума на криво? 3, рис. VIII.3) не является квазистационарным. Однако, через некоторое вреьа после достижения максимума режим реакции приближается к квазистационарному. Покажем это.
Зависимость концентраций исходного вещества А и промежуточного продукта Р от времени для реакции (Х.99) имеет вид
Если переписать это ура
ра легко видеть, что при h >i [ахувеличивается со врем:енг«_ асуществлен.
Но при ki <§ck2 можно наш выражение (Х.110) перехолйг! » е. квазистационарный рев) •снованный на использована* Котекании реакции в закрытой ■вих концентраций.
Если реакция состоит аз ■ ронних и двусторонних), то каш :ько при определенных усэа ГЛ. Последовательность олвв Диавионарвов, есш. констант £стант скоростей всех оспин \ 2. Последовательность jvafl шонарной, если она включает < ■намного меньше констант саване Нам направлениях.
г 3. Последовательность, вся Нвв, будет квазистационарвов» ■ваших стадий будет значатся односторонних стадий.
{ _ В качестве примера рассмотри* |
гме:
с а—сме
-kit.
(Х.106"
Ijcrb нам известно, что константа.
СР=СА0
(Х.1(П
г..
k2-k
Условие квазистационарности в рассматриваемом случае запишется в вид;
Следовательно, согласно вмени будет протекать квалисгашнав] иых веществ В, С и D:
340
 -=1.
-=1.
конечные отклонения сР от :млемое время. Условимся кнтрацию Ср=0,95сР(СТ). Тогдь
dcp a *1
— = *i"CA - К2Ср(ст) = О? Ср(ст) =~~ СА,
dt k2
re сд:— концентрация вещества А в данный момент времени L Подставляя значение сА из (X. 106) в (Х-108), находим
(ХЛ08)
ср=сА0
—е
*2
3
(Х.105
О-3 с, т. е. время достижения
Щ) при постоянстве концент-стационарных концентраций
закрытой системе концент-иться постоянными, так как d временем. Однако для реак-понятие квазистационарного нцентрации промежуточны?, (ремени отвечают условиям концентрациям исходных точных и исходных веществ жменимо и для квазистацио-
DHapHoro режима требование Епродолжительности их жиз-иие, чем при стационарном промежуточных частиц смо-исходного вещества в каж-кбуемые при стационарном
состояния для модельной крытой системе, т. е. когда со временем. Кинетику этой чного вещества меняется со (до максимума на кривой гнако, через некоторое время ближается к квазистационар-
»а А и промежуточного про-
(Х.106)
'). (х.107)
сом случае запишется в виде
.-сюда
Если переписать это уравнение в виде
(/1_^-1),
Ср *2 , ttj-fcjV
(X.1Q9) (Х.110)
(Х.1 И)
Ср(ст) *2
то легко видеть, что при kt>k2 Отношение Щ/сц^] при f >imax больше единицы и увеличивается со временем, т. е. квазиравновесный режим не может быть осуществлен.
Но при fci«rk2 можно пренебречь величиной кх в разности к2~к\ и тогда выражение (X. 110) переходит в условие (Х.104) для стационарности процесса, т. е.: квазистационарный режим может быть реализован. Метод расчета, основанный на использовании условия (Х.98) для промежуточных веществ при протекании реакции в закрытой системе, называется методом квазистационарных концентраций.
; Если реакция состоит из нескольких последовательных стадий (односторонних и двусторонних), то квазистационарный режим может быть достигнут только при определенных условиях.
Последовательность односторонних (необратимых) стадий будет квазистационарной, если константа скорости первой стадии значительно меньше констант скоростей всех остальных стадий.
Последовательность двусторонних (обратимых) стадий будет квазистационарной, если она включает одну стадию, Обе константы скорости которой намного меньше констант скоростей всех Остальных стадий в прямом и обратном направлениях.
Последовательность, включающая двусторонние и односторонние стадии, будет квазистационарной, если константа скорости первой из односторонних стадий будет значительно меньше констант скоростей остальных односторонних стадий.
_ В качестве примера рассмотрим многостадийную реакцию, протекающую в закрытой системе:
кх кг к, kt
А ДгВ Z? C-*D-»E (Х.112)
(Х.113)
Пусть нам известно, что константа скорости к% значительно меньше других:
k3<Kki, k_i, к2, fc_2, к*.
Следовательно, согласно условию 3, эта реакция в закрытой системе начиная с некоторого времени будет протекать квазистационарно. Запишем условия квазистационарности для промежуточных веществ В, С и D:
341
(Х.Ш
(Х.115
(Xllf
(Х1Г
(Х.Ш
(X.11J
"kicA+к-гсс—i св—к2св щ О,
dcc
<1св 17'
dec
17
i &2CB — focc — к&С ** 0»
=k3cc-kAcD-0.
fcl(fc-2+*3)CA £_1(£-2+*э)+м:з
Из уравнений (X.115) и (X.114)
*2Св
ее*-——; св =
fc_l(fc_24-fc3)-l-fe2fc3
а:1^2^зсА
и, следовательно, из (Х.117) и (Х.116)
ее
*з
Cd=— Сс=—
fej fc( i (fc_2 + £3) + £2&з]
Чтобы получить выражение кинетической кривой для вещества А (т. е. зависимость са от t в явне* виде), учтем, что при протекании реакции в квазистационарном режиме скорости всех стада! практически одинаковы и равны скорости реакции в целом. Рассмотрим первую и четверг)"-: стадии:
1 dcA 1 dco
= к^Сц.
(-1) dt (-1) dt
deA
кцСо
Из этого соотношения с учетом (Х.119) получаем
dt
к\к2късА
(Х.12
fc4[fc-i (аг-г+^зн^з]
Интегрируя это уравнение
dcA &i&2*3
Учитывая, кроме того, .119) к виду
Ср.-
= сдо{-1-|
рве К\ и К2 —константы w можно получить сразу, предаст^ Зри этом К\ — св/сд, K2—ccJct ш МЩ
■ввшненЕЯ (Х.123) в эти выражшшж, a
Такое приближение назьяв ■не кинетической задачи да я лни, протекающей в квазясжяй простых алгебраических урняя
Если механизм прс константы скорости отде ства достаточно активны (i
довательно, находятся в ре [то часто для упрощения реакции принимают, что В этом случае метод: как приближенный мете к кинетическим уравнениям, т мерности реального процесса
АО"
dCA k„i(k-2 + k3) + k2k3
In-
■t
получаем зависимость концентрации исходного вещества А от времени:
СА кХк2кЪ
сА0 fc_i(fc_2+fc3)+fc2fc3
Главе XI
теоретические предстал химической кинетики