

7.Синдром вторичного индуцированного иммунодефицита
Система иммунобиологического надзора (ИБН) представляет собой набор
факторов противоинфекционной резистентности и форм иммунного ответа, ко-
торые защищают организм от патогенных микробов, благодаря чему большин-
ство инфекций протекает кратковременно и без нарушающих здоровье послед-
ствий. Совокупность этих факторов, называющихся иммунитетом, выполняет
функцию защиты организма от генетически чужеродных веществ (антигенов)
экзогенного или эндогенного происхождения с целью сохранения гомеостаза,
структурной и функциональной целостности организма, а также биологической
(антигенной) индивидуальности. Иммунный ответ заключается в распознавании
возбудителя и инициировании цепи реакций, направленных на его устранение.
Все разнообразные формы иммунореактивности подразделяются на два типа:
врожденные (неспецифическая резистентность) и приобретенные (адаптивные)
иммунные реакции.
Врожденный (естественный, наследственный, неспецифический) иммунитет
является первичной воспалительной реакцией организма и направляет неспеци-
фические факторы защиты на нейтрализацию и выведение патогенов. Он вклю-
чает в себя:
— неспецифическое распознавание патоген-ассоциированных молекулярных
паттернов (микробных антигенов) посредством паттерн-распознающих рецепто-
ров семейства TLR (Toll-like receptor), имеющихся на поверхности макрофагов,
нейтрофилов, дендритных клеток и В-лимфоцитов;
— фагоцитоз, осуществляемый моноцитами/макрофагами и нейтрофилами; — факторы неспецифической защиты, к которым относятся система компле-
мента (лектиновый и альтернативный пути активации), антимикробные белки
(кателицидины, дефензины, лизоцим, фибронектин, белки острой фазы), естест-
венные антитела [IgM] (иммуноглобулин М ) и цитокины.
Клетки системы врожденного иммунитета, активированные через рецепторы
TLR, синтезируют и секретируют провоспалительные цитокины и хемокины,
такие как IL-1β, IL-6, IL-8, TNFα и интерфероны 1 типа (IFN-α и IFN-β), кото-
рые участвуют в развитии приобретенного иммунитета.
Ведущая роль в реакциях приобретенного (адаптивного, специфического) им-
мунитета принадлежит лимфоцитам, которые специфически взаимодействуют
с конкретными антигенами. В-лимфоциты продуцируют антитела (иммуно-
глобулины), специфически связывающие определенные молекулы-мишени — ан-
тигены. Различные популяции Т-лимфоцитов выполняют различные функции.
Т-лимфоциты фенотипа Th1 (Т-хелперы типа 1) участвуют в иммунном воспале-
нии, активируя макрофаги, и стимулируют клеточный иммунитет. Т-лимфоциты
фенотипа Th2 (Т-хелперы типа 2) участвуют в регуляции гуморального иммуни-
тета (дифференцировки В-лимфоцитов и продукции антител). Цитотоксические
Т-лимфоциты с помощью антигенраспознающего рецепторного комплекса
TCR/CD8 участвуют в специфическом распознавании пептидов антигенного про-
исхождения с последующим уничтожением (киллингом) инфицированных внут-
риклеточным патогеном клеток или опухолевых клеток-мишеней.
Связь неспецифических и специфических механизмов ИБН осуществляется
посредством антигенпрезентирующих клеток (АПК), к которым относятся мак-
рофаги и дендритные клетки. АП Кпоглощают и процессируют (расщепляют)
чужеродные антигены до антигенных пептидов, а затем связывают их с молеку-
лами главного комплекса гистосовместимости (major histocompatibility complex,
МНС), у человека он обозначается HLA (human leukocyte antigens), и презенти-
руют наивным Т-лимфоцитам. Молекулы МНС класса I презентируют эндоген-
ные антигены (антигены вирусов, опухолевых клеток и другие) цитотоксиче-
ским Т-лимфоцитам, имеющим на своей поверхности антигенраспознающий ре-
цепторный комплекс TCR/CD8. Молекулы МНС класса II презентируют
антигены экзогенной природы Т-хелперам с антигенраспознающим рецептор-
ным комплексом TCR/CD4. Антигенраспознающие рецепторы TCR (Т cell recep-
tor) одного лимфоцита распознают только один антиген. После формирования
комплекса TCR/антиген-пептид/МНС класса I или II активированный Т-лимфо-
цит пролиферирует с образованием клона такой же специфичности, как и исход-
ный Т-лимфоцит. Активированные Т-лимфоциты секретируют растворимые ре-
гуляторные факторы (цитокины), которые активируют соответствующий эф-
фекторный механизм иммунного ответа. Причем Th1 и Th2 оказывают друг на
друга супрессирующее действие: клетки Th1 продуцируют IFN-γ, который угне-
тает Th2, а Th2 продуцируют IL-10, который угнетает Th1. Выбор эффекторного
механизма, зависящий от баланса Th1/Th2, очень важен, так как от него зависит
адекватность иммунного ответа.
Система комплемента — это одна из систем врожденного иммунитета, функ-
ция которого состоит в том, чтобы отличать «свое» от «не-своего». Такая диф-
ференциация осуществляется благодаря присутствию на клетках организма ре-
гуляторных молекул, подавляющих активацию комплемента. Компоненты ком-
племента представляют собой растворимые медиаторы иммунитета,
сывороточные белки, которые находятся в неактивном состоянии и последова-
тельно активируются при наличии в организме чужеродного антигена в следую щем порядке: C1q, C1r, C1s, C4, C2, C3, C5, C6, C7, C8, C9. Существуют три
основных пути активации комплемента — классический, альтернативный и лек-
тиновый. При классической (антителозависимой) активации комплемента про-
исходит связывание иммунных комплексов антиген–антитело с компонентом
комплемента C1q, что соединяет врожденный иммунитет (комплемент) с приоб-
ретенным (антитела).
Кроме компонентов комплемента, к растворимым медиаторам иммунитета
относятся цитокины (интерфероны, интерлейкины, хемокины, колониестимули-
рующие факторы и др.), посредством которых осуществляется межклеточная
сигнализация при развитии воспалительного процесса. На его начальных стади-
ях местные иммунные клетки могут секретировать провоспалительные цитоки-
ны IL-1 и IL-6. Активированные лимфоциты и макрофаги, появляющиеся в оча-
ге воспаления, тоже выделяют цитокины IL-1, TNF, IL-4, IFN-γ. Другие цитоки-
ны и хемокины (хемотаксические цитокины), например IL-8, оказывают
хемотаксическое и активирующее действие на иммунные клетки, мигрирующие
из кровотока в очаг воспаления.
При нарушении функции одного или нескольких элементов иммунной систе-
мы (недостаточности или избыточности) развиваются иммунопатологические
состояния, к которым относятся иммунодефициты, патологическая толерант-
ность, аллергия, болезни аутоиммунной агрессии, реакция «трансплантат про-
тив хозяина».
Иммунодефицитные состояния или иммунологическая недостаточ-
ность — это нарушение иммунного ответа, сопровождающееся развитием реци-
дивирующих инфекций и опухолей. Различают первичные (врожденные) и вто-
ричные (приобретенные) иммунодефициты.
Первичные иммунодефициты развиваются вследствие генетических де-
фектов (мутаций) в иммунных клетках и проявляются в первые месяцы или го-
ды жизни. При первичных ИДС, связанных с иммуноглобулинами, компонента-
ми комплемента и фагоцитозом (хроническая гранулематозная болезнь, дефи-
цит молекул адгезии лейкоцитов, дефицит миелопероксидазы, синдром
гипериммуноглобулинемии Е (синдром Джоба) и синдром Чедиака — Хигаси),
наблюдается повышенная чувствительность к повторным пиогенным инфекци-
ям, вызываемым гноеродными бактериями, обладающими капсулой и вызываю-
щими гнойное воспаление, — Haemophilus influenzae, Streptococcus pneumoniae
и Staphylococcus aureus. При первичных иммунодефицитных состояниях, связан-
ных с нарушениями клеточного иммунитета (функций Т-лимфоцитов), повыша-
ется чувствительность к оппортунистическим микроорганизмам, безвредным
для здоровых людей. У индивидов с недостаточностью Т-клеточной функции
они могут вызывать генерализованные инфекции.
Первичные гуморальные иммунодефициты (нарушения В-клеточного звена
иммунитета) выражаются в нарушениях антителообразования. Кним относятся
сцепленная с Х-хромосомой агаммаглобулинемия, селективный дефицит IgA,
селективный дефицит субклассов IgG, гипер-IgM-синдром, общий вариабель-
ный иммунодефицит, транзиторная гипоглобулинемия детского возраста.
Сцепленная с Х-хромосомой агаммаглобулинемия развивается только у маль-
чиков в результате мутации гена Btk, кодирующего В-клеточную тирозинкиназу
Брутона, которая является компонентом сигнального каскада, активирующего
В-лимфоциты. Данная мутация блокирует их дифференцировку на стадии
пре-В-лимфоцитов. В лимфоузлах пациентов отсутствуют герминативные цент-
ры и плазматические клетки, а в крови — зрелые В-лимфоциты, иммуноглобу- лины IgA, IgM, IgD и IgE, количество IgG понижено. Отсутствуют антитела к ан-
тигенам АВО. Эффективна заместительная терапия (внутривенное введение им-
муноглобулина 1 раз в неделю или нативной плазмы 1 раз в месяц).
Селективный дефицит IgA возникает в результате дефектов хромосомы 18
и проявляется снижением (или отсутствием) IgA на фоне нормального содержа-
ния других иммуноглобулинов. Лица с недостаточностью IgA подвержены забо-
леваниям, в развитии которых играют роль иммунные комплексы (гиперчувст-
вительность III типа), а также инфекционным, аллергическим и аутоиммунным
осложнениям. Появление антител против IgA может вызвать анафилактические
реакции. Лечение симптоматическое.
Селективный дефицит субклассов IgG (дефицит IgG2 и/или IgG4 при нормаль-
ном уровне IgG) возникает вследствие нарушения терминальной дифференци-
ровки В-клеток и проявляется повышенной чувствительностью к рецидивирую-
щим инфекциям пиогенных микроорганизмов. Рекомендована иммунизация боль-
ных с рекуррентными инфекциями дыхательных путей конъюгированной вак-
циной против Haemophilus influenzae или Streptococcus pneumoniae, при которой
происходит переключение синтеза антител: вместо IgG2, образующихся против
полисахаридных антигенов, синтезируются IgG1, образующиеся против белко-
вых антигенов. В случае тяжелого течения инфекции показано введение донор-
ских иммуноглобулинов.
Гипер-IgM-синдром характеризуется дефицитом IgG и IgA в сочетании с ги-
перпродукцией IgM. В 70 % случаев эта форма иммунодефицита развивается толь-
ко у мальчиков вследствие мутации в гене, локализованном на Х-хромосоме и
кодирующем белок CD40L (CD154) активированных Т-лимфоцитов. Белок
CD40L участвует во взаимодействии Т-лимфоцитов с В-лимфоцитами и денд-
ритными клетками, экспрессирующими белок CD40. Дефицит CD40L блокирует
дифференцировку В-лимфоцитов и переключение синтеза иммуноглобулинов
с IgM на IgG, IgA и IgE. Описаны аутосомные варианты гипер-IgM-синдрома,
обусловленные мутациями в генах транскрипционного фактора NF-κB и цити-
диндезаминазы. Гипер-IgM-синдром проявляется увеличением лимфоузлов
и селезенки и часто осложняется аутоиммунными заболеваниями (гемолитиче-
ской анемией, тромбоцитопенической пурпурой и др.). Для лечения назначают
иммуноглобулины и антимикробные препараты.
Общий вариабельный иммунодефицит (ОВИД) проявляется снижением
уровня одних иммуноглобулинов при нормальном содержании других (дисгам-
маглобулинемия), гиперплазией лимфоузлов и лимфоидного глоточного кольца
и подверженностью рецидивирующим инфекциям дыхательных путей и желу-
дочно-кишечного тракта. Патогенетические механизмы ОВИД разнообразны:
дефекты В-лимфоцитов или Т-хелперов CD4+, нарушение взаимодействия
Т-лимфоцитов с В-лимфоцитами вследствие мутации гена CD154, дефицит ци-
токинов, дефект генов МНС класса III (комбинированный иммунодефицит).
У многих больных наблюдается спленомегалия и/или диффузная лимфаденопа-
тия, развиваются аутоиммунные заболевания (особенно злокачественная ане-
мия) и лимфомы. При этом количество циркулирующих В-лимфоцитов не изме-
няется. Для предупреждения повторных гнойных инфекций больные с ОВИД
должны в течение всей жизни получать внутривенно гаммаглобулин.
Транзиторная гипоглобулинемия детского возраста — исчезновение IgG, по-
лученного плодом через плаценту от матери, вскоре после рождения. Количест-
во IgG и IgA в сыворотке крови при транзиторной гипоглобулинемии снижено,
а IgM — в норме или повышено. В-клетки детей с транзиторной гипоглобулине- мией не имеют дефектов, вероятно, они не получают достаточного активирую-
щего сигнала от Т-лимфоцитов CD4+. Лечение обычно не проводится, детям
с частыми инфекционными осложнениями назначают заместительную иммуног-
лобулинотерапию.
Первичные иммунодефициты с генетическими нарушениями Т-клеточного
звена иммунитета проявляются как комбинированный или тяжелый комбини-
рованный иммунодефицит. Так как В-лимфоциты функционально зависят от
Т-клеточных сигналов, мутационные дефекты Т-лимфоцитов проявляются как
комбинированная недостаточность и клеточного, и гуморального иммунитета.
Тяжелые комбинированные иммунодефициты (ТКИД) составляют 40 % всех
первичных иммунодефицитов. Х-сцепленный ТКИД развивается только у маль-
чиков вследствие мутаций гена IL2RG (локус Xq13), кодирующего γ-цепь рецеп-
торов IL-2 и других цитокинов (IL-4, IL-7, IL-9, IL-11, IL-15, IL-21), регулирую-
щих дифференцировку и функционирование Т-лимфоцитов. При этом отсутст-
вуют Т-лимфоциты и NK-клетки и снижен синтез иммуноглобулинов. Лечение
состоит в трансплантации костного мозга или генотерапии путем введения гена
IL2RG в гематопоэтические стволовые клетки.
Аутосомно-рецессивные ТКИД обусловлены мутациями в генах ADA (адено-
зиндезаминаза), JAK3 (сигнальная тирозинкиназа, передающая сигнал с цитоки-
новых рецепторов), IL-7RА (α-цепь рецептора IL-7), RAG1 и RAG2 (рекомбина-
зы — белки реаранжировки генов иммуноглобулинов), Artemis (фермент репа-
рации ДН Кпосле реаранжировки иммуноглобулиновых генов рекомбиназами
RAG1 и RAG2), CD45 (трансмембранный белок лейкоцитов со свойствами тиро-
зинфосфорилазы, регулирует сигнальную трансдукцию в лимфоцитах). Пациен-
ты со всеми формами ТКИД получают симптоматическое лечение и/или транс-
плантацию костного мозга. В стадии разработки находятся генотерапевтические
подходы по замещению мутантного гена в Т-лимфоцитах «здоровым» геном.
В отличие от ТКИД, комбинированные Т-клеточные иммунодефициты ха-
рактеризуются наличием у пациентов минимальной Т-клеточной активности.
Кним относятся недостаточность пуриннуклезидфосфорилазы, возникающая
вследствие мутации гена PNF, наследственная атаксия-телеанги-эктазия вслед-
ствие мутации гена АТМ (синдром Луи — Бар), недостаточность антигенов МНС
классов I и II (синдром «голых лимфоцитов»), нарушение активации Т-лимфо-
цитов вследствие мутации гена ZAP-70, кодирующего зета-цепь сигнальной ти-
розинкиназы, синдром Ди Джорджи (гипоплазия тимуса) вследствие мутаций в
локусах DGCR (DiGeorge syndrome chromosome region, хромосома 22) и DGCR2
(хромосома 10), Х-сцепленный синдром Вискотта — Олдрича (мутация в гене
WAS, локус Хр11.22–11.23). При всех этих синдромах наблюдается Т-клеточная
недостаточность. Лечение, как правило, симптоматическое, так как специфиче-
ская терапия в большинстве случаев не разработана. Возможна трансплантация
костного мозга и генотерапия (трансфекция здоровых генов в стволовые клетки
или Т-лимфоциты больных).
Вторичные (приобретенные иммунодефициты) развиваются под влия-
нием повреждающих факторов (болезней, нарушений обмена веществ, истоще-
ния, авитаминоза, стрессов, травм) и иммуномодулирующих лекарственных пре-
паратов. Наиболее важными лекарственными препаратами, используемыми для
системной иммунотерапии, являются стероиды, которые оказывают выражен-
ное влияние на многие стадии и компоненты иммунного ответа, миграцию и
функции иммунных клеток, синтез цитокинов, изменяют популяционный состав
циркулирующих лейкоцитов. Применение стероидов вызывает лимфоцитопе-
нию, моноцитопению и нейтрофилию. Стероиды ингибируют активацию и про-
лиферацию лимфоцитов, особенно Т-лимфоцитов. Результатом подавления про-
дукции провоспалительных цитокинов является угнетение клеточных и гумораль-
ных иммунных процессов вследствие подавления функции Т-хелперов.
Иммуномодулирующим действием обладают циклофосфамид, хлорамбуцил,
азатиоприн, циклоспорин, рапамицин, метотресат и другие цитостатики. Меха-
низмы иммуносупрессирующих эффектов лекарственных препаратов различны.
Они могут угнетать пролиферацию и активность иммунных клеток, антигенпре-
зентирующую функцию моноцитов.
Вторичный иммунодефицит может развиться под влиянием дефицита неко-
торых элементов питания. Например, недостаток цинка снижает соотношение
CD4+/CD8+ и нарушает функции Т-клеток, недостаток железа нарушает проли-
ферацию лимфоцитов и фагоцитирующую способность нейтрофилов. Дефицит
витамина А снижает численность некоторых субпопуляций лимфоцитов, недо-
статок витамина В6 и фолата нарушает клеточный иммунитет.
Синдром приобретенного иммунодефицита (СПИД) — тяжелое потенциаль-
но летальное инфекционное заболевание, возбудителем которого является вирус
иммунодефицита человека (ВИЧ). От СПИДа погибает 2—3 млн человек в год.
Инфекция характеризуется длительным латентным периодом (у половины ин-
фицированных терминальная стадия развивается в течение 9—10 лет) и разно-
образием клинических проявлений. ВИЧ-1 и ВИЧ-2 (human immunodeficiency
virus — HIV-1 и HIV-2) относятся к семейству Retroviridae, роду Lentivirus. ВИЧ-2
эндемичен для Африки и менее патогенен.
ВИЧ содержит двуцепочечную РНК. Основные гены ВИЧ: gag (коровый бе-
лок), pol (ретротранскриптаза) и env (белок оболочки). ВИЧ инфицирует преи-
мущественно Т-лимфоциты CD4+, так как его рецептором является антиген CD4,
с которым связывается вирусный гликопротеин gp120. Инфицируются также
макрофаги, дендритные клетки и клетки мозга. В процессах интернализации ви-
руса участвуют хемокиновые ко-рецепторы CXCR4 Т-лимфоцитов и CCR5 мак-
рофагов. Хроническая инфекция не сопровождается клиническими проявления-
ми. Со временем у половины инфицированных индивидов развиваются истоще-
ние и дисфункция субпопуляции Т-лимфоцитов CD4+, что является основной
причиной иммунодефицита при ВИЧ-инфекции. Инфицированные клетки ста-
новятся недоступными для ВИЧ-специфичных цитотоксических Т-лимфоцитов,
так как вирус подавляет в них экспрессию молекул МНС класса I. Уровень сыво-
роточных IgG повышен, в результате неспецифической поликлональной актива-
ции В-лимфоцитов появляются неэффективные «бессмысленные» глобулины.
На стадии СПИДа у больных отмечаются неспецифические симптомы общего
характера, вирусные, бактериальные и грибковые инфекции кожи и слизистых,
развиваются тяжелые оппортунистические инфекции и опухоли, особенно часто
саркома Капоши (многоочаговая опухоль эндотелиального происхождения, ас-
социированная с вирусом герпеса 8), а также В-клеточные лимфомы, ассоцииро-
ванные с вирусом Эпштейна — Барр.
Лечение СПИДа основано на антиретровирусных препаратах — нуклеозид-
ных и ненуклеозидных ингибиторах ретротранскриптазы, протеазы и интегразы
вируса. Энфувиртид блокирует слияние вируса с мембраной клетки. Хемоки-
ны — лиганды хемокиновых рецепторов, ко-рецепторов CD4 — подавляют ин-
+
фицирование Т-лимфоцитов CD4 . Цитокины IFN-α и IL-12 ингибируют репро-
дукцию ВИЧ. Кроме того, проводят лечение, направленное на контроль инфек-
ционных осложнений у больных. Разрабатываются вакцины против ВИЧ.
Гемостаз. Понятие о первичной и вторичной реакции гемостаза. Механизмы антитромбогенных свойств эндотелия.
Система гемостаза — это биологическая система в организме, функция которой заключается в сохранении жидкого состояния крови, остановке кровотечений при повреждениях стенок сосудов и растворении тромбов, выполнивших свою функцию. Различают три основных механизма остановки кровотечения при повреждении сосудов, которые в зависимости от условий могут функционировать одновременно, с преобладанием одного из механизмов:
Сосудисто-тромбоцитарный гемостаз, обусловленный спазмом сосудов и их механической закупоркой агрегатами тромбоцитов. На обнажившихся в результате повреждения стенки сосуда коллагеновых молекулах происходит адгезия (прилипание), активация и агрегация (склеивание между собой) тромбоцитов. При этом образуется так называемый «белый тромб», то есть тромб с преобладанием тромбоцитов.
Коагуляционный гемостаз (свертывание крови), запускается тканевым фактором из окружающих повреждённый сосуд тканей, и регулируемый многочисленнымифакторами свертывания крови. Он обеспечивает плотную закупорку повреждённого участка сосуда фибриновым сгустком — это так называемый «красный тромб», так как образовавшаяся фибриновая сетка включает в себя клетки крови эритроциты. Раньше сосудисто-тромбоцитарный гемостаз называли первичным, коагуляционный вторичным, так как считалось, что эти механизмы последовательно сменяются, в настоящее время доказано, что они могут протекать независимо друг от друга.
Фибринолиз — растворение тромба после репарации (ремонта) повреждённой стенки сосуда.
Конечным итогом работы свертывающей системы крови является превращение фибриногенав волокнафибринапод действиемтромбина. Установлено, что любой сгусток, который образуется в сосудах, в том числе в артериях, является тромбоцитарно-фибриновым. Тромбоциты играют важную роль в восстановлении стенок сосуда: из тромбоцитов, участвующих в образовании сгустка, выделяется большое количество активных веществ. В числе прочих выделяетсяфактор роста тромбоцитов(англ.Platelet-derived growth factor, PDGF) — сильный стимулятор восстановления тканей. Завершающий этап работы системы гемостаза — фибринолиз. Система фибринолиза разрушает фибриновый сгусток по мере того, как повреждённый сосуд восстанавливается, и необходимость в наличии сгустка пропадает.
Гемостаз - это система обеспечения и сохранности жидкого состояния крови и ОЦК путем предупреждения и остановки кровотечений. Биологическое значение этой системы - поддержание гомеостаза путем обеспечения нормального кровоснабжения органов и сохранения необходимого ОЦК.
Структурно-функциональные основы гемостаза представлены в основном тремя взаимодействующими между собой компонентами:
Стенкой кровеносных сосудов, в первую очередь ее интимой.
Клетками крови.
Плазменными ферментными системами: свертывающей, антисвертывающей (фибринолитической).
При патологии гемостаза основным клиническим проявлением является кровоточивость.
19.2. Понятие о первичной и вторичной реакции гемостаза
До недавнего времени решающее значение в осуществлении гемостаза приписывалось свертывающей системе крови. Однако современные исследования показали, что на повреждение кровеносных сосудов первыми реагируют сами сосуды (спазм, открытие шунтов выше места повреждения) и клетки крови (тромбоциты и отчасти эритроциты). Известно также, что тромбоцитам, а не свертыванию крови принадлежит ведущая роль в первичной остановке кровотечений из микрососудов. Время кровотечения из мельчайших сосудов крови всегда удлинено при тромбоцитопениях и тяжелых дисфункциях кровяных пластинок и остается нормальным при гемофилиях и многих других нарушениях свертываемости крови. Вследствие этих причин первичный гемостаз - это сосудисто-тромбоцитарная реакция на потерю крови, а вторичная гемостатическая реакция - это реакция свертывания крови (РСК). Хотя оба эти механизмы включаются не строго последовательно друг за другом, они на значительном отрезке времени функционируют одновременно и сопряженно.
19.2.1. Начальный этан - сосудисто-тромбоцитарный гемостаз
Стенки кровеносных сосудов играют важную роль не только в обеспечении структурной основы гемостаза, но и в поддержании жидкого состава крови. Интима сосудов, эндотелий обладает очень высокой тромборезистентностью, в силу чего сохранность этой внутренней выстилки - важнейшее условие сохранения жидкого состояния крови. В основе этой тромборезистентности лежат сложные и пока далеко не полностью расшифрованные механизмы.
Ингибиторы гемостаза:
а) антисвертывающие - эндотелий синтезирует и секретирует мощный ингибитор агрегации тромбоцитов и расширения микрососудов - простациклин;
б) эндотелий синтезирует и секретирует основной физиологический антикоагулянт - антитромбин III, без которого гепарин не активен.
в) эндотелий фиксирует на своей поверхности с помощью специальных рецепторов гепарин и активный комплекс гепарин-антитромбин III,
г)эндотелий вырабатывает и выделяет в кровоток мощные активаторы фибринолиза - это белковые активаторы плазминогена, синтезируемые в сосудистой стенке.
Активаторы гемостаза:
а) коллаген, активирует XII фактор РСК, запускающий свертывание. Коллаген одновременно реализует запуск не только свертывания крови, но и фибринолитические (антисвертывающие) системы. По мере заживления раны происходит рассасывание тромба и восстановление микроциркуляции;
б) участие тромбоцитов в гемостазе определяется в основном следующими функциями этих клеток:
ангиотрофической, т.е. способностью поддерживать нормальную структуру и функцию стенок микрососудов [показать]
адгезивно - агрегационной, т.е. способностью тромбоцитов приклеиваться (адгезия) к поврежденным участкам сосудистой стенки и быстро образовать в таких местах тромбоцитарную пробку (агрегация), останавливающую кровотечение [показать]
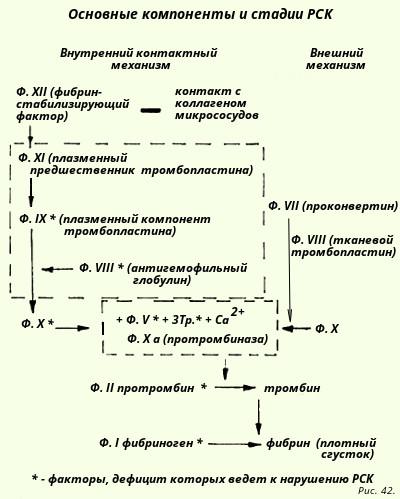
19.2.2. Вторичная гомеостатическая реакция - реакция свертывания крови
Основы современной ферментной теории свертывания крови были заложены в прошлом столетии А.А.Шмидтом и уточнены Моравитцем. Согласно этой теории, образование волокон фибрина, составляющих каркас любого свертка крови, связано с ферментным oтщеплением от молекул фибриногена небольших фрагментов, после чего остающиеся основные части этих молекул (фибрин-мономеры) соединяются друг с другом в длинные цепи (фибрин-полимеры). Фермент крови, вызывающий отщепление фибринопептидов и превращение фибриногена в фибрин, получил название тромбина.
Имеется два различных механизма активации реакции свертывания крови: "внутренний" и "внешний" .
"Внутренний" механизм запускается в результате контакта XII фактора РСК с коллагеном поврежденного сосуда, в результате запускается серия реакций, завершающаяся образованием X фактора.
"Внешний" механизм - тканевой тромбопластин образуется при интенсивном разрушении клеток и представляет собой фосфолипиды клеточных мембран, содержащие Са2+. Запуск серии реакций также ведет к образованию X фактора. Отличие: участвуют меньше факторов и быстрее идет РСК. Далее за образованием X активного фактора идут общие этапы реакции свертывания крови.
Т.о., схематично процесс свертывания крови можно представить как трехфазный процесс:
I фаза - образование X фактора (различен при внешнем и внутреннем механизмах);
II фаза - образование тромбина (общий);
III фаза - формирование фибрина (общий);
Основные показатели реакции свертывания крови:
Количество тромбоцитов - норма 180-350 х 109 л.
Время свертывания цельной крови - норма 5-11 минут.
Протромбиновое время (тест Квика - определяют время свертывания рекальцифицированной плазмы при добавлении к ней стандартного количества тканевого тромбопластина; отражает содержание в плазме II, VII, X факторов). Норма - 12 сек., допускается 15-16 сек.
19.3. Основные виды патологии гемостаза
19.3.1. Тромбоцитопении [показать]
19.3.2. Тромбоцитопатии [показать]
19.3.3. Нарушения коагуляционного гемостаза:
наследственные нарушения свертывания крови (гемофилии):
приобретенные геморрагические коагулопатии:
дефицит К-витаминозависимых факторов;
синдром диссеминированного внутрисосудистого свертывания крови (ДВС-синдром).
19.3.3.1. Наследственные нарушения свертывания крови
Гемофилия - семейно-наследственный геморрагический диатез, связанный с нарушением 1-й фазы РСК (образования плазменного тромбопластина). Заболевание обусловлено отсутствием или недостаточным содержанием в крови одного из белков (глобулинов) плазмы, принимающих наряду с Са и фактором III тромбоцитов участие в образовании кровяного тромбопластина.
В зависимости от вида дефицитного фактора различают следующие виды гемофилий. Гемофилия А - дефицит антигемофильного глобулина фактора VIII встречается в 70-78% случаев всех гемофилий. Болезнь Виллебранда - в основе болезни лежит нарушение синтеза основного крупномолекулярного компонента фактора VII в эндотелии кровеносных сосудов (единственном месте синтеза его в организме). Нарушение синтеза фактора IX - ведет к гемофилии В (болезнь Кристмаса), фактора XI - к гемофилии С (фактора XI - плазменный предшественник тромбопластина, активатор фактора IX).
Гемофилией, как правило, болеют мужчины. Заболевание может передаваться по наследству практически здоровыми женщинами, так называемыми "кондукторами". Но в настоящее время описаны 7 случаев так называемой "женской" гемофилии, при этом только женщинами наследуется легкая форма дефицита фактора VIII. Выделяют семейные и спорадические формы болезни.
Патогенез всех видов гемофилий связан с замедленным и недостаточным образованием фактора Ха, что, в свою очередь, ведет к недостаточному образованию тромбина и, следовательно, фибрина. В результате процесс свертывания крови резко замедляется, поэтому повреждение любого, даже самого мелкого сосуда, осложняется длительным кровотечением.
Клиника: тяжесть заболевания зависит от степени дефицита белков реакции свертывания крови. У некоторых "кондукторов" уровень фактора VIII в плазме может составлять 11-12%, что создает угрозу кровотечения при травмах, операциях, родах. Об этой опасности следует помнить при хирургических вмешательствах у родственников мужчин, больных гемофилией. У них следует проверить фактор VIII. Основными симптомами заболевания являются кровоизлияния в суставы (гемартрозы), подкожные и межмышечные гематомы, возникающие после даже незначительной травмы, а также длительные кровотечения при нарушении целостности кожи и слизистой оболочки (случайные порезы, экстракции зубов).
Лабораторные пробы: прежде всего отмечается резкое удлинение времени свертывания цельной крови и активированного парциального тромбопластинового времени. Прогноз в отношении жизни в основном является благоприятным, с возрастом клинические проявления болезни смягчяются, поскольку сознательное устранение травмирующих факторов позволяет избежать кровотечений.
Лечение: в период геморрагических обострений необходимо внутривенное введение гемопрепаратов, содержащих фактор VIII (антигемофильная плазма - криопреципитат, выделяемый из плазмы с помощью охлаждения и содержащий белковый концентрат, в котором достаточно фактора VIII, фибриногена и фактора XIII, но мало альбумина и других белков).
Помнить! При лечении существует опасность переноса вируса СПИДа, сывороточного гепатита.
19.3.3.2. Приобретенные геморрагические коагулопатии
Дефицит К-витаминозависимых факторов. Этот синдром характеризуется нарушением синтеза в гепатоцитах и, следовательно, снижением концентрации в плазме Са2+ - содержащих белков РСК: VII - проконвертина, IX -антигемофильного глобулина В, X фактора Пауэра-Стюарта, II - протромбина.
Различают следующие патогенетические варианты этого синдрома:
Недостаточность образования витамина К в кишечнике (при геморрагической болезни новорожденных, при энтеропатиях, дисбактериозе медикаментозного генеза в результате лечения антибиотиками широкого спектра).
Заболевания печени, ведущие к:
а) недостаточному всасыванию жирорастворимого витамина К при нарушении переваривания липидов в кишечнике из-за отсутствия желчи в кишечнике (механическая желтуха);
б) при нарушении белковой функции печени (гепатиты, циррозы и т.д.) - нарушается синтез в печени плазменных факторов свертывания VII, X, IX, У, XI, I и ингибиторов фибринолиза.
При применении и передозировке функциональных антагонистов витамина К (применение антикоагулянта косвенного механизма действия).
Синдром диссеминированного внутрисосудистого свертывания крови.
ДВС - синдром иначе называется тромбогеморрагическим синдромом и представляет собой распространенный и потенциально опасный вид патологии гемостаза, в основе которого лежит рассеянное свертывание крови в циркуляторном русле с образованием множества микросгустков и агрегатов клеток крови, блокирующих кровообращение в органах и вызывающих в них циркуляторную гипоксию и дистрофические изменения.
Этиология: при интенсивном разрушении клеток фосфолипиды мембран клеточных органелл, содержащие Са2+ (это и есть тканевой тромбопластин - активатор РСК ) попадают в кровоток. Так происходит при одонтогенном сепсисе с фагоцитозом и последующей гибелью белых клеток, некрозе опухолей в результате ишемии, действии цитотоксических ядов или излучений, преждевременной отслойке плаценты, абортах или задержке мертвого плода в полости матки, сильных черепно-мозговых травм. В ряде случаев так происходит при внутрисосудистых повреждениях, например, иссечении аневризмы, множественных укусах насекомых. Одновременно из лизосом разрушенных клеток в кроветок попадает активатор анти РСК - системы фибринолиза. Последствия: множественное тромбообразование с активацией фибринолиза. Проявляется в виде венозной тромбоэмболии и кровотечения с места травмы, хирургического вмешательства.
Патогенез состоит из двух фаз, сменяющих друг друга:
I фаза - гиперкоагуляция. Происходит распространенное, рассеянное (диссеминированное) свертывание крови главным образом в русле микроциркуляции. При этом образуются множественные тромбы, которые разносятся по кровотоку и оседают в паренхиматозных органах. Развивающаяся блокада тромбоэмболами русла микроциркуляции приводит к недостаточной перфузии, что следует причиной нарушения функции этих органов и описывается термином "шоковый" орган.
II фаза - гипокоагуляция. Множественное внутрисосудистое свертывание крови приводит к значительному потреблению факторов коагуляции. При этом продукция факторов коагуляции не успевает за их потреблением. Следствием является компенсаторный гиперфибринолиз, активируемый и появлением антикоагулянтных продуктов расщепления фибриногена. В терминальном периоде процесс гипокоагуляции может доходить до возникновения полной несвертываемости крови. При этом возникают кровотечения, не поддающиеся терапии.
Суть ДВС - на фоне диссеминированного тромбообразования человек погибает от кровотечения в месте травмы во 2-й фазе или в 1 фазе от тромбоэмболии.
Диагностика ДВС - резкое удлинение времени свертывания крови, протромбинового времени, тромбоцитопения до 9 х 104 клеток/л, снижение содержания фибриногена.
Патогенетические принципы терапии ДВС:
в 1 фазу - введение антикоагулянтов (гепарин).
во 2 фазу - введение коагулянтов: свежая плазма крови, тромбоцитарная взвесь.
Существует опасность вызвать стимуляцию 1 фазы - тромбообразования.
Профилактика осуществляется путем введения за несколько часов до операции гепарина. Гепарин, блокируя все три фазы реакции свертывания крови, способен оборвать эту цепную реакцию и тем самым предотвратить развитие геморрагических осложнений: в 1-й фазе РСК - образование X фактора (тромбопластина), во 2 фазе - образование тромбина, в 3-й фазе - образование фибрина.
Все вещества, секретируемые эндотелием и участвующие в гемостазе и тром-
бозе, можно, в известной степени условно, разделить на две группы: тромбоген-
ные и атромбогенные (табл. 10.2).
Таблица 10.2
Тромбогенные и атромбогенные факторы сосудистой стенки
Тромбогенный фактор Звено гемостаза Атромбогенный фактор
Фактор Виллебранда (vWF), тром- Тромбоцитарно- Протеаза, разрушающая фактор
боксан А (TxA ), фактор актива- сосудистое звено Виллебранда (ADAMTS-13), про-
2 2
ции тромбоцитов (PAF), АДФ гемостаза стациклин (PGI ), простагландин
2
Е (PGE ), оксид азота (NO), моно-
2 2
окись углерода (CO), экто-АДФаза
Тканевой фактор Коагуляционное Ингибитор тканевого пути сверты-
звено гемостаза вания (TFPI), тромбомодулин, про-
теогликаны
Ингибитор активатора плазмино- Фибринолиз Тканевой активатор плазминогена
гена (PAI-1) (t-PA), урокиназный активатор
плазминогена (u-PA)
Квеществам, индуцирующим адгезию и агрегацию тромбоцитов, относятся
фактор Виллебранда (ФВ), фактор активации тромбоцитов (РАF), аденозинди-
фосфорная кислота (АДФ), тромбоксан А (ТхА ). Адгезия тромбоцитов к эндо-
2 2
телию и субэндотелиальному матриксу — начальный этап гемостаза и тромбоза.
В норме адгезия тромбоцитов к неповрежденному эндотелию происходит в очень
ограниченном объеме и необходима для обеспечения капилляротрофической
функции. В условиях патологии адгезия ограничивается, как правило, зоной
прилежащей к области повреждения сосудистой стенки. За пределами зоны по-
вреждения эндотелия адгезия тромбоцитов ограничена образованием эндотели-
альными клетками простациклина, оксида азота (NO), экто-АДФазы и других
факторов, ингибирующих адгезию и агрегацию тромбоцитов.
Адгезия и агрегация тромбоцитов приводит к образованию тромбоцитарного
тромба, который в условиях нормальной функции эндотелия прочно связан
с сосудистой стенкой. Этот этап гемостаза связан с активацией плазменных про-
коагулянтов и образованием тромбина — фактора, вызывающего необратимую
агрегацию тромбоцитов, а также ключевого фермента системы свертывания
крови, под влиянием которого фибриноген превращается в фибрин. Тромбин,
кроме того, является активатором эндотелиоцитов. Из эндотелия в условиях по-
вреждения выделяется тканевой фактор (ТФ), инициирующий внешний (быст-
рый) путь свертывания крови. Ингибиторы образования тромбина (ингибитор
тканевого фактора TFPI), тромбомодулин, протеогликаны и др.) предотвращают
избыточное фибринобразование на луминальной поверхности сосудов при по-
вреждении сосудистой стенки, а также (вместе с плазменными ингибиторами
тромбиногенеза) драматическое внутрисосудистое свертывание крови. Наконец,
в эндотелии образуются активаторы и ингибиторы фибринолиза — процесса,
имеющего большое значение в «судьбе» тромба.
При всем разнообразии в строении и механизмах действия тромбогенных и
атромбогенных факторов есть общие закономерности в их образовании и учас-
тии в гемостазе и тромбозе. Многие из этих факторов, образующихся в эндоте- лии, выполняют функцию тромборегуляторов. Тромборегуляторы не являются
веществами строго специфичными с точки зрения их образования и действия;
они оказывают влияние не только на гемостаз, но и на другие процессы: прони-
цаемость, вазомоторные реакции (простациклин, NО, ТХА ), ангиогенез, кле-
2
точную пролиферацию (тканевой активатор плазминогена) и т. д. Источниками
тромборегуляторов при определенных условиях могут быть лейкоциты, макро-
фаги, клетки опухолей и другие клетки.
Тромборегуляторы эндотелиального происхождения, имеющие сравнительно
большой период биологического полураспада, оказывают не только локальное,
но и системное действие на клетки крови и кровеносные сосуды. Это относится,
прежде всего, к тканевому фактору, простациклину, тканевому активатору плаз-
миногена и его ингибитору. Вещества, секретируемые эндотелием, оказывают как
прямое влияние на гемостаз (ФВ, тромбомодулин и др.), так и опосредованное
(эндотелин 1 и др.). В регуляции гемостатической функции эндотелия большое
значение имеют гормоны (вазопрессин, эстрогены), цитокины (интерлейкин-1 —
ИЛ-1, фактор некроза опухолей альфа — ФНО-α), гемодинамические факторы.
В физиологических условиях образование атромбогенных веществ в эндоте-
лии преобладает над образованием тромбогенных, что обеспечивает сохранение
жидкого состояния крови при повреждениях сосудистой стенки, в том числе не-
значительных, случайных, которые могут иметь место в норме. Секреция атром-
богенных веществ обеспечивает тромборезистентность кровеносных сосудов.
При функциональных нагрузках на сосуды и при активации эндотелия обра-
зование и выделение оксида азота, простациклина, активатора плазминогена и,
возможно, других факторов тромборезистентности возрастает, и это есть прояв-
ление неспецифической реакции. Это связано с тем, что регуляция образования
атромбогенных факторов, некоторые из которых являются и вазодилататорами
(простациклин, оксид азота), во многом зависит от гемодинамических факторов.
В настоящее время сформировалось представление о наличии в эндотелии «ме-
ханосенсоров», которые располагаются на поверхности эндотелиоцита, в цито-
скелете, в местах межклеточных соединений. При увеличении напряжения сдви-
га развиваются быстрые (< 1 мин) реакции (выделение NO, PGI2) и медленные
(1—6 часов) реакции (увеличение образования NO-синтазы, t-РА, ТФ, тромбо-
модулина и других факторов). В механизме быстрых реакций большое значение
имеют активация калиевых каналов (в течение миллисекунд), изменение кон-
центрации Са2+, гиперполяризация мембраны эндотелиоцита, активация G-бел-
ков. Медленные реакции связаны с увеличением синтеза тромборегуляторов
(t-PA, PAI-1), а также эндотелиальной NO-синтазы. Поскольку напряжение
сдвига и другие гемодинамические факторы влияют на эндотелий артерий
в большей степени, чем на эндотелий вен, в артериальных сосудах образование
атромбогенных веществ превышает таковое в венозных сосудах. В норме атром-
богенные вещества сосудистой стенки не препятствуют гемостазу при поврежде-
нии сосудов, но ограничивают процесс тромбообразования; в этом и заключает-
ся значение тромборезистентности.
При повреждении эндотелия нередко происходит уменьшение образования
атромбогенных веществ и увеличение образования тромбогенных факторов, что
является риском развития тромбоза. Поскольку тромборезистентность стенки
артериальных сосудов значительно выше, чем тромборезистентность стенки ве-
нозных сосудов, и это различие сохраняется даже в условиях патологии, тром-
боз артерий встречается значительно реже, чем тромбоз вен.
9.Методы оценки системы гемостаза
Система гемостаза - биологическая система, обеспечивающая, с одной стороны, сохранение жидкого состояния циркулирующей крови, а с другой – предупреждение и купирование кровотечений.
Компоненты системы гемостаза:
сосудисто-тромбоцитарное звено
система свертывания крови (коагуляция)
физиологические антикоагулянты
фибринолитическая система (тромболизис)
1.1. Сосудисто-тромбоцитарный гемостаз
В сосудисто-тромбоцитарном механизме свертывания крови участвуют сосуды, ткань, окружающая сосуды и форменные элементы крови (главная роль принадлежит тромбоцитам). Тромбоциты образуются в костном мозге из мегакариоцитов. Продолжительность их жизни около 9 суток. При недостаточном количестве тромбоцитов или их функциональной неполноценности развивается микроциркуляторный тип кровоточивости. К важнейшим функциям тромбоцитов относят адгезивно-агрегационную и ангиотрофическую. В условиях нормы эндотелий эффективно предупреждает процессы адгезии, агрегации тромбоцитов, а также реакций коагуляции. Способность эндотелия сохранять кровь в жидком состоянии обеспечивается синтезом ингибитора агрегации тромбоцитов простациклина и отрицательным зарядом эндотелиальных клеток. Кроме того, эндотелиальный белок тромбомодулин препятствует уже начавшейся коагуляции. Основной функцией тромбомодулина является инактивация тромбина и превращение (модификация) его в мощный активатор антикоагулянтной системы - протеин С. За счет этого происходит значимое снижение скорости коагуляционных реакций. Эндотелий участвует в фибринолизе за счёт синтеза и выделения в кровоток тканевого плазминогенового активатора, который активирует плазминовую систему. При повреждении мелкие сосуды спазмируются. Этот спазм обусловлен сокращением гладкомышечных клеток, он возникает рефлекторно и продлевается серотонином, тромбоксаном А2, катехоламинами и другими вазоконстрикторами, которые появляются из эндотелиальных клеток и тромбоцитов. Повреждение сосудов сопровождается быстрой активацией тромбоцитов. Эта активация обусловлена появлением высоких концентраций АДФ (из поврежденных эритроцитов и сосудов), а также появлением коллагеновых и фибриллярных структур из субэндотелия. Контакт крови с коллагеном немедленно ведёт к адгезии тромбоцитов, реализуемой с участием рецепторов GP-Ia, GP-Ib и фактора Виллебранда. Под влиянием АДФ, тромбоксана А2 и катехоламинов тромбоциты склеиваются между собой, образуя агрегаты, которые являются основой тромбоцитарной пробки. Усилению агрегации способствует тромбин, всегда появляющийся в результате свертывания крови в месте повреждения. Агглютинация и агрегация сопровождается изменением формы тромбоцитов и появлению рецепторов на мембране тромбоцитов к фибриногену (GPIIb-IIIa), благодаря чему, в присутствии ионов Са++, последний связывает между собой активированные тромбоциты. Такая связь между активированными тромбоцитами не прочна. Именно поэтому такую агрегацию называют обратимой. Образование прочной тромбоцитарной пробки следует после вторичной агрегации, которая сопровождается секрецией из тромбоцитов ПгG2, ПгH2, тромбоксана А2, ионов Са++, фактор активации тромбоцитов (ФАТ), адреналина, норадреналина, фибриногена и многих других. Секреция этих веществ обусловлена активацией актомиозиновой системы тромбоцитов, что обуславливает выделение вышеперечисленных субстанций из тромбоцитов за счёт повышения давления внутри тромбоцита. Кроме того, активация актомиозиновой системы ведет к ретракции (сокращению и уплотнению) тромбоцитарной пробки. В норме кровотечение из мелких сосудов прекращается не более чем через 5 минут.
1.2. Коагуляционный гемостаз
При повреждении крупных кровеносных сосудов тромбоцитарная пробка не способна остановить кровотечение. Только коагуляционный гемостаз способен остановить кровотечение из крупного сосуда. В коагуляционных реакциях принимают участие специальные белки, фосфолипиды (из тромбоцитарной мембраны), ионы кальция. Большинство белков, участвующих в коагуляции, являются проферментами (обозначаются римскими цифрами). Их активация осуществляется за счет протеолиза (они обозначаются римскими цифрами с добавлением буквы а, например, IIа, Xа, Vа и др.).
1.2.1. Международная номенклатура факторов свертывания крови
|
Название фактора |
Количество в мл (активность) |
Достаточный минимум |
Период полужизни |
Избыток |
|
I. Фибриноген |
300 (170-450) мг |
50 мг |
100 ч. |
3-6 раз |
|
II. Протромбин* |
200мкг/70-130% |
80 мкг/40% |
72 - 96 ч |
2-3 раза |
|
III. Тромбопластин |
- |
- |
- |
- |
|
IV. Ионы Са++ |
2,3 - 2,8 ммоль/л |
- |
- |
- |
|
V. АС-глобулин |
25мкг/80-110% |
2,5-4мкг/10-15% |
12 - 15 ч. |
8-10 раз |
|
VII. Проконвертин |
2 мкг/70-130% |
0,2 мкг/10% |
2 - 6 ч. |
10 раз |
|
VIII. Антигемофильный глобулин |
50мкг/80-120% |
5-7мкг/10-15% |
7 - 8 ч. |
3-5 раз |
|
IX. Кристмас-фактор |
3-4 мкг/70-130% |
4-6мкг/20-30% |
20 - 30 ч. |
4-5 раз |
|
X. Стюарта-Прауэра фактор* |
6-8 мкг/70-140% |
0,15мкг/20% |
30 - 70 ч. |
5 раз |
|
XI. Предшественник тромбопластина |
7 мкг/70-130% |
15 мкг/15-20% |
30 - 70 ч. |
4-5 раз |
|
XII. Хагеманна фактор |
40 мкг |
не установлено |
50 - 70 ч. |
неизвестно |
|
XIII. Фибриназа, фибрин-стабилизирующий фактор |
не установлено |
10% |
72 - 100 ч |
10 раз |
* синтезируется в печени
Витамин"К"-зависимые факторы: II, VII, IX, X Чувствительные к тромбину факторы: I, V, VIII, XIII Факторы контакта: XII, XI, BM-кининоген, прекалликреин Факторы-сериновые протеазы: XII,XI,X,IX,VII, II, Плазмин Дополнительные факторы:
Фактор Виллебранда
Фактор Флетчера
Фактор Фитцжеральда
Процесс свертывания крови - это целая цепь последовательных ферментативных реакций, в которой проферменты, активируясь, способны активировать другие факторы свертывания крови. Удобно рассматривать схему коагуляции в виде каскада ферментативных реакций, условно разделенного на внутренний и внешний механизмы. Конечным продуктом коагуляционных реакций и по внешнему и по внутреннему механизму является фибрин.
1.2.2. Схема свертывания крови [А.Н. Мамаев 2003]
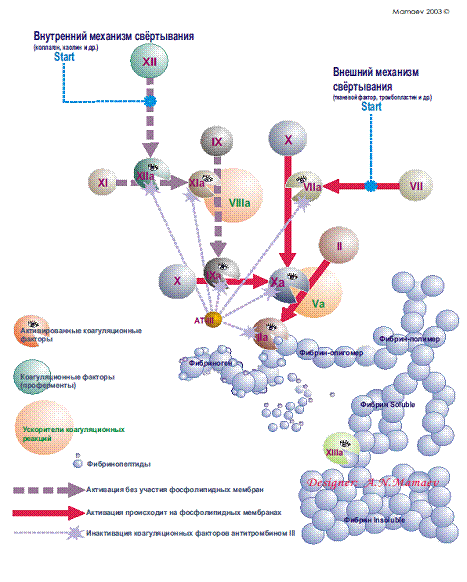
Внешний механизм коагуляции Внешний механизм свертывания предполагает обязательное наличие тканевого фактора (фактора III), а старт коагуляции начинается с активациифактора VII. Активированный фактор VII переводит фактор X в Xа и активирует фактор IX (активация фактора IX идет медленно и существенной роли в коагуляции не играет). Затем фактор Xа переводит протромбин (II) в тромбин. Эту реакцию значительно ускоряют коагуляционный фактор Vа и фосфолипиды. Образование фибрина инициализируется по внешнему пути очень быстро (в течение секунд), что ведет к появлению первых порций тромбина, активирующих другие коагуляционные факторы (VIII, V, XIII и др.).
Внутренний механизм коагуляции Старт коагуляции по внутреннему механизму начинается с активации фактора Хагемана (XII) и происходит на фосфолипидных мембранах тромбоцитов. Фактор Хагемана активируется коллагеном из эндотелия, адреналином и др., а затем уже активированная молекула фактора Хагемана преобразует фактор XI в XIа. В этой реакции принимает участие калликреин, который также активируется фактором XIа. В свою очередь, фактор XIа активирует фактор IX. Фактор IXа на фосфолипидных мембранах с участием фактора VIIIа и ионов Са++ путем протеолиза превращает фактор X в его активированную форму. Далее фактор Xа переводит протромбин в тромбин. Эту реакцию значительно ускоряют коагуляционный фактор Vа и фосфолипиды.
Конечный этап коагуляции Переход фибриногена в фибрин происходит следующим образом: от фибриногена тромбин отщепляет 2 фибринопептида А и 2 фибринопептида В. Так образуются фибрин-мономеры. Затем формируются димеры, тримеры и олигомеры фибрина. После этого образуются фибриллы растворимого фибрина. Фибрин-стабилизирующий фактор (активированный тромбином) в присутствии Са++ превращает нестабильный, растворимый фибрин в стабильный нерастворимый фибрин. В результате этого сгусток фибрина становится резистентным к фибринолитическим агентам и с трудом разрушается другими протеолитическими веществами. Образовавшийся сгусток фибрина уплотняется за счет тромбоцитов, в большом количестве попадающих в структуру сгустка. Наступает ретракция сгустка фибрина. Сгусток, состоящий из тромбоцитов, эритроцитов и большого числа волокон фибрина, способен остановить кровотечение из крупных сосудов.
1.3. Физиологические антикоагулянты
Физиологические антикоагулянты разделяют на первичные и вторичные. Первичные антикоагулянты всегда присутствуют в крови, а вторичные образуются в результате коагуляционных реакций.
К первичным антикоагулянтам относятся:
антитромбин III;
протеин С;
протеин S;
ингибитор внешнего пути свертывания (TFPI);
кофактор гепарина II.
Одним из основных антикоагулянтов является антитромбин III (АТ). Антитромбин III обладает мощным антикоагулянтным действием только в комплексе с гепарином. Этот комплекс надежно блокирует коагуляционные факторы IIа, IXа, Xа, XIа, XIIа и калликреин. Дефицит АТ – серьезный фактор риска развития венозных тромбозов. Другим ингибитором свертывания является кофактор гепарина II. Его действие усиливается во много раз при взаимодействии с гепарином. Однако клиническая значимость его невелика. К антикоагулянтам относится ингибитор внешнего пути свертывания (TFPI). Установлено, что он тормозит образование фактора Xа по внешнему механизму коагуляции. Антикоагулянтная система протеина С включает в себя целую цепь последовательных биохимических реакций. Образующийся в процессе коагуляции тромбин связывается на эндотелии с мембранным гликопротеином – тромбомодулином, и вследствие этого теряет всю свою коагуляционную активность, но сохраняет способность активировать протеин С. После этого активированный протеин С с протеином S в качестве кофактора, на фосфолипидной поверхности расщепляет фактор Vа и фактор VIIIа. Этот механизм эффективно предупреждает дальнейшее образование тромбина и трансформирует его в активатор антикоагулянтного механизма.
Вторичными антикоагулянтами являются продукты деградации фибриногена и фибрина. Они тормозят конечный этап коагуляции.
1.4. Система фибринолиза
Фибриновый сгусток, образовавшийся в результате свертывания крови, в дальнейшем подвергается лизису под влиянием ферментов фибринолитической системы крови, происходит восстановление проходимости сосудов. Кроме того, фибринолитическая система контролирует заживление ран и выполняет ряд других важных функций.
Фибринолиз включает 4 компонента:
основной фермент – плазмин;
плазминоген (неактивный предшественник плазмина);
активаторы плазминогена;
ингибиторы плазминогена.
Активация плазминогена может происходить по внешнему и внутреннему механизму [10].
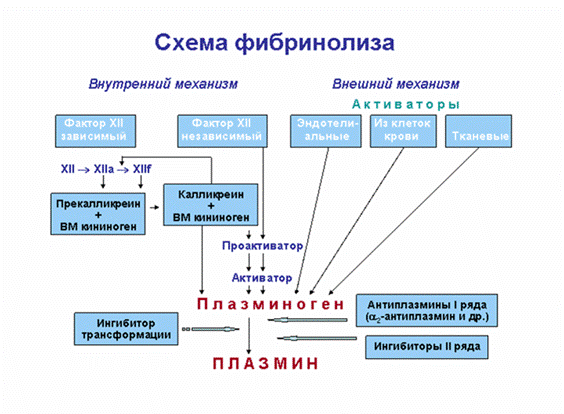
Основным активатором внешнего механизма является тканевый активатор плазминогена, синтезирующийся в эндотелиальных клетках, и урокиназа.Внутренняя активация осуществляется преимущественно комплексом ф.XIIa с калликреином (так называемый XIIa - зависимый фибринолиз). Фибринолиз может быть двух видов: первичный и вторичный. Первичный фибринолиз вызывается гиперплазминемией, при поступлении в кровь большого количества активаторов плазминогена. Вторичный фибринолиз развивается в ответ на внутрисосудистое свертывание крови, вызванное поступлением в кровоток тромбопластических веществ. Активаторы плазминогена преобразуют плазминоген в плазмин, а последний вызывает протеолиз фибрина. В результате протеолиза в кровотоке появляются продукты деградации фибрина (ПДФ). Важнейшими ингибиторами фибринолиза являются антиплазмины I ряда - ПАИ-1, ПАИ-2 и ?2-антиплазмин. Менее значимы ингибиторы II ряда – ?2-макроглобулин, антитрипсин, антитромбин III и С1-ингибитор. Большое клиническое значение имеет определение в крови одного из ПДФ, а именно D-димера, так как этот показатель является наиболее надежным маркёром образования фибрина внутри сосуда.
Основные осложнения патологии гемостаза:
кровотечение (при тромбоцитопении или дисфункции тромбоцитов, болезни Виллебранда, гемофилии А (В), клинической манифестации ДВС);
внутрисосудистое свертывание крови (артериальные, венозные и смешанные тромбозы обусловленные тромбофилией или без нее, ДВС-синдром (острый, подострый, хронический), тромботическая тромбоцитопеническая пурпура).
Диагностика патологии гемостаза направлена на решение следующих задач:
определение причин различных видов кровоточивости и тромбозов, подбор специфических методов профилактики и лечения;
отбор групп риска для предупреждения послеоперационных кровотечений и тромбоэмболий;
снижение летальности и инвалидизации при неотложных и критических состояниях, протекающих с ДВС-синдромом;
решение проблем привычного невынашивания беременности при антифосфолипидном синдроме (АФС), тромбофилиях;
контроль безопасности и эффективности терапии антикоагулянтами, антиагрегантами, тромболитиками, средствами заместительной терапии.
2.Методы исследования системы гемостаза
Клинико-функциональные пробы при исследовании сосудисто-тромбоцитарного звена системы гемостаза (в условиях нашей лаборатории не проводятся):
определение ломкости микрососудов с помощью пробы манжеточной компрессии (проба Кончаловского-Румпель-Лееде); определение времени кровотечения из микрососудов без дополнительной компрессии (проба Дьюка с проколом уха и др.), либо на фоне венозного стаза (сдавление плеча манжетой до 40 мм.рт.ст. с проколами или надрезами кожи предплечья) – пробы Айви и Борхгревинка и др.
Лабораторные методы:
измерение числа и функции тромбоцитов (адгезия, агрегация) путем микроскопии или с использованием гематологических анализаторов (при скрининговых исследованиях) и агрегометров;
функциональные коагуляционные, или так называемые клоттинговые (по оценке времени свертывания мануально или с использованием коагулометров разных конструкций);
определение параметров фибринолиза;
амидолитические (тесты с использованием хромогенных субстратов к тромбину, плазмину, фактору Xа, XIIIа и др., и фотометров с фиксированной длиной волны измерений);
иммунологические методы, позволяющие выявить уровень искомого антигена или антител при АФС и др.
выявление генетических аномалий методом ПЦР (мутации Лейден-резистентности фактора Vа к активированному протеину С, гена протромбина G 20210, гена метилентетрагидрофолатредуктазы и др.).
Для улучшения качества исследования системы гемостаза важно придерживаться следующих принципов. При обследовании больных необходимо выделять два последовательных этапа диагностики: первичного скрининга с использованием скрининговых тестов и – на втором этапе – проб, позволяющих уточнить диагноз. Для подтверждения диагноза в случае выявления серьезных нарушений в системе гемостаза (снижение уровня фактора Виллебранда, факторов свертывания, тромбоцитопении, дефицита или аномалии действия физиологических антикоагулянтов, наличия волчаночного антикоагулянта, выраженной тромбинемии и др.) необходимо повторное обследование. Интерпретация показателей коагулограммы должна проводиться с учетом возможного влияния принимаемых лекарственных средств и других воздействий. Например, учитывать особенности питания при контроле за лечением антикоагулянтами непрямого действия (АНД). Целесообразно отказаться от дублирующих или малоценных, а также устаревших и неточных методов исследования (приложение 1). Использование производительных и высокоточных (по сравнению с мануальными определениями) коагулометров и агрегометров, а также стандартизированных расходных материалов, является предпочтительным. В данном пособии приведены референсные значения, которые приняты в нашей лаборатории. Референсные величины могут варьировать при смене лота или производителя набора, актуальные референсные значения указываются в бланке результата анализа.
3. Тесты для оценки сосудисто-тромбоцитарного компонента гемостаза
При тромбоцитопениях, тяжелых тромбоцитопатиях и при дефиците фактора Виллебранда (ФВ) значительно удлиняется время кровотечения. Кровоточивость связана с недостаточностью адгезивно-агрегационной функции тромбоцитов – нарушением образования в поврежденных сосудах тромбоцитарной пробки. Это может быть обусловлено либо значительным снижением количества тромбоцитов в крови, либо их дисфункцией, в основе которой чаще всего лежит отсутствие или блокада на мембране тромбоцитов рецепторов, взаимодействующих со стимуляторами (агонистами) агрегации этих клеток (ФВ, адреналином, АДФ, фибриногеном, арахидоновой кислотой и простагландинами), либо отсутствием в тромбоцитах или нарушением выхода из них компонентов гранул, содержащих эти стимуляторы агрегации. Качественные дефекты тромбоцитов, лежащие в основе большого числа геморрагических диатезов, подразделяют на следующие группы:
дезагрегационные тромбоцитопатии, обусловленные отсутствием или блокадой мембранных рецепторов этих клеток (тромбастения Гланцмана и др.);
болезни отсутствия плотных и ?-гранул;
нарушения высвобождения гранул;
нарушения образования циклических простагландинов и тромбоксана А2;
дефицит, аномалии и нарушения мультимерности ФВ;
нарушения обмена нуклеотидов и транспорта кальция.
3.1. Время кровотечения
Время кровотечения - это время от момента нанесения стандартной раны кожи до момента прекращения вытекания крови. Оно характеризует функциональную активность тромбоцитов и взаимодействие тромбоцитов с сосудистой стенкой. Время кровотечения не выявляет всех тромбоцитарных нарушений (такого метода вообще не существует), этот скрининговый тест позволяет заподозрить тромбоцитопатии различного генеза, болезнь Виллебранда и нарушения проагрегантных свойств сосудистой стенки. После выявления патологии нет необходимости повторять это исследование, нужно использовать более чувствительные и специфические методы. У этого метода есть серьезные недостатки:
Метод плохо стандартизируется. Результаты теста позволяют лишь предположить наличие тех или иных нарушений.
Низкая чувствительность. Отсутствие удлинения времени кровотечения не всегда позволяет исключить нарушения тромбоцитарного или сосудистого звеньев гемостаза.
Низкая специфичность не позволяет однозначно интерпретировать результаты метода.
Не соответствует современным санитарно-эпидемиологическим требованиям.
3.2. Количество тромбоцитов в крови
Референсные значения: 170-350x109/л
СНИЖЕНИЕ ЧИСЛА тромбоцитов (<170?109/л):
острый ДВС-синдром;
острый лейкоз и миелодиспластические синдромы;
гипо- и апластические анемии;
нарушение образования в организме тромбоцитопоэтина;
химиотерапия и лучевая терапия;
тромботическая тромбоцитопеническая пурпура и гемолитико-уремический синдром;
спленомегалия и гепатолиенальный синдром;
гепарин-индуцированная тромбоцитопения;
эклампсия и преэклампсия;
экстракорпоральное кровообращение;
гемодиализ у больных с хронической почечной недостаточностью, гемосорбция;
интенсивная трансфузионная терапия;
пароксизмальная ночная гемоглобинурия;
иммунные формы патологии (СКВ и др. коллагенозы, АФС, иммунная тромбоцитопеническая пурпура);
дефекты при получении крови для исследования - псевдотромбоцитопения в случае использования ЭДТА в качестве стабилизатора крови.
ПОВЫШЕНИЕ ЧИСЛА тромбоцитов (>350?109/л):
мегакариоцитарные и миелолейкозы, эритремия;
вторичный, реактивный тромбоцитоз в случае спленэктомии (через 1-3 недели), внутриполостные кровоизлияния после оперативных вмешательств, спустя 7-10 дней от начала подострого токсико-инфекционного ДВС-синдрома, после перенесенного острого кровотечения, при злокачественных новообразованиях (предвестник опухоли легкого, поджелудочной железы) и других причинах хронического ДВС-синдома.
3.3. Индуцированная агрегация тромбоцитов
При исследовании функций тромбоцитов индуктор агрегации добавляется к плазме, обогащенной тромбоцитами. Для исследования индуцированной агрегации тромбоцитов используют физиологические индукторы, такие как тромбин, адреналин, АДФ, коллаген. Кроме того, существуют специальные индукторы, такие как ристоцетин (ристомицин). Этот индуктор инициирует связывание фактора Виллебранда с мембранным рецептором Ib-IX тромбоцитов и таким образом вызывает их агрегацию. Для диагностики большинства наследственных и приобретенных тромбоцитопатий достаточно исследования функциональных параметров тромбоцитов с использованием четырех агонистов. Ими являются индукторы АДФ, адреналин, коллаген и ристомицин. Исследование агрегации на стекле менее чувствительно, чем с использованием агрегометра, однако быстро выполняется и используется при скрининге для отбора пациентов с грубыми нарушениями тромбоцитарного гемостаза (выраженной тромбоцитопенией или тромбоцитопатией) в группу риска для профилактики, например, интра- и послеоперационных кровотечений.
Методы определения агрегации тромбоцитов:
с АДФ
с адреналином
с коллагеном
с арахидоновой кислотой
с тромбином
с ристомицином (ристоцетином)
Большие преимущества имеет графическая регистрация процесса на агрегометре, однако выполнение исследований требует большого количества плазмы и затрат времени. Агрегометры подразделяются на оптические (турбодиметрические), регистрирующие агрегацию в богатой тромбоцитами плазме по изменению её оптической плотности, и кондуктометрические, определяющие агрегацию в цельной крови по изменению электропроводности. Результаты этих исследований позволяют диагностировать тромбоцитопатии, нозологическая принадлежность которых обусловлена характерным нарушением тех или иных функциональных свойств тромбоцитов или их сочетанием (см. таб. 1).
Таблица 1. Изменение агрегатограмм при нарушениях функции тромбоцитов
|
Состояние |
АДФ |
Адрена-лин |
Арахидо-новая кислота |
Тромбин |
Коллаген |
Ристоми-цин |
|
Референсные значения: |
8-12 с |
15-20 с |
|
|
15-20 с |
|
|
Болезнь Виллебранда |
Н |
Н |
Н |
Н |
Н |
v |
|
Синдром Бернара-Сулье |
H |
H |
H |
H/v |
H |
v |
|
Тромбастения Гланцмана |
v |
v |
v |
v |
v |
+/- |
|
Передозировка аспирина |
v |
v |
v |
+/- |
v |
+/- |
|
Синдром серых тромбоцитов |
v |
v |
H/v |
+/- |
v |
+/- |
H – нормальная агрегатограмма, v - сниженная реакция на индуктор агрегации
ПОВЫШЕНИЕ АГРЕГАЦИОННОЙ АКТИВНОСТИ тромбоцитов характерно для претромботических состояний, идиопатического тромбоцитоза, тромбозов, инфарктов органов, атеросклероза, васкулитах, при беременности.
СНИЖЕНИЕ АГРЕГАЦИИ наблюдается при первичных и симптоматических тромбоцитопатиях, при лечении антиагрегантами. Антиагреганты отличаются по механизму действия. Одни антиагреганты (аспирин, нестероидные противовоспалительные средства) блокируют образование в тромбоцитах простагландиновых стимуляторов агрегации, в частности тромбоксана А2, другие ингибируют АДФ-рецепторы (клопидогрель), третьи нарушают транспорт ионов кальция в тромбоциты либо стимулируют образование циклического аденозинмонофосфата. Классификация современных антиагрегантов приведена в таблице 2.
Таблица 2. Ингибиторы сосудисто-тромбоцитарного гемостаза [3]
|
Группы препаратов в соответствии с механизмом действия |
Лекарственные препараты |
|
Ингибиторы циклооксигеназы (СОХ-1). Основной механизм: блокада образования циклических простагландинов |
Аспирин (кардиомагнил, тромбоас), другие нестероидные противовоспалительные средства (индометацин и др.) |
|
Ингибиторы тромбоксансинтетазы |
Сулотробан и др. |
|
Ингибиторы тромбоксансинтетазы и тромбоксановых рецепторов |
Пикотамид, ридогрель и др. |
|
Блокаторы тромбиновых рецепторов тромбоцитов |
Ванипрост, дальтробан. |
|
Блокаторы АДФ-рецепторов тромбоцитов |
Тиенопиридины: тиклопидин (тиклид), клопидогрель (плавикс). |
|
Антагонисты рецепторов IIb/IIIa тромбоцитов |
Антительные: абсиксимаб (Reo Pro). Пептидные: интегрилин и др. Непептидные: тирофибан, ламофибан. Оральные антагонисты рецепторов: имлофибан, фрадафибан. |
|
Стабильные производные простациклина |
Инъекционные формы: илопрост, вазопростан. Оральные формы: берапрост. |
|
Препараты комплексного действия и вазопротекторы |
Пентоксифиллин (трентал), сульфин-пиразоны, дипиридамол (курантил), эндотелон, миртравен. |
4.Скрининговые тесты для оценки плазменного звена гемостаза
Лабораторная диагностика нарушений системы гемостаза является одной из самых дорогостоящих в лабораторной практике. Выполнение всех возможных тестов для уточнения характера нарушений для всех пациентов – практически нереальная задача. Поэтому чрезвычайно важно соблюдать этапность проведения тестов, исходить из клинических данных и анамнеза пациента. На первом этапе для уточнения направленности нарушений необходимо провести тесты, отражающие состояние целых звеньев системы гемостаза. Поскольку в разных лабораториях при анализе гемостаза преследуются разные цели, перечень тестов, входящих в гемостатический скрининг для данной лаборатории, может отличаться от такового в других лабораториях. Однако существует набор рекомендуемых тестов, традиционно называемых скрининговыми для диагностики состояния системы гемостаза.
Скрининговые тесты:
АПТВ (активированное парциальное тромбопластиновое время
протромбиновое время (по Квику)
тромбиновое время и/или фибриноген
Скрининговые тесты на состояние внутреннего и внешнего каскада активации протромбиназы позволяют выявлять нарушения со стороны факторов-субстратов, кофакторов, ингибиторов каскада свертывания, а также действие некоторых лекарственных препаратов или аутоантител. Основным тестом на состояние внутреннего каскада свертывания плазмы является АПТВ, на состояние внешнего каскада - протромбиновое время.
4.1. Активированное парциальное (частичное) тромбопластиновое время (АПТВ)
АПТВ используется как скрининговый тест для оценки внутреннего каскада свертывания плазмы, скрининговой диагностики волчаночного антикоагулянта и слежения за антикоагулянтным действием гепаринов. АПТВ – более значимый тест для первичного выявления патологии, чем протромбиновое время, так как выявляет относительно часто встречающуюся гемофилию А и В (дефицит факторов VIII и IX соответственно) и наличие волчаночного антикоагулянта.
Референсные значения АПТВ: 28,6-33,6 с
УКОРОЧЕНИЕ АПТВ:
активация внутреннего механизма свертывания при тромбозах, тромбоэмболиях. Это может быть связано с резистентностью фактора V к активированному протеину С, повышенным уровнем фактора VIII или активированных факторов свертывания;
при ДВС-синдроме (гиперкаогуляционная фаза);
возможно при нормально протекающей беременности.
УДЛИНЕНИЕ АПТВ:
дефицит факторов внутреннего пути свертывания (VIII - гемофилия А, IX – гемофилия В, XI, XII) при нормальных результатах протромбинового теста;
дефицит факторов II, V, X в случае сопутствующей гипокоагуляции в протромбиновом тесте;
дефицит фактора Виллебранда;
гепаринотерапия обычным, нефракционированным гепарином (НГ) (тест выявляет сравнительно низкие концентрации антикоагулянта, приблизительно от 0,05 МЕ/мл крови);
лечение антикоагулянтами непрямого действия (АНД);
ДВС-синдром (потребление факторов свертывания в фазу гипокоагуляции);
на фоне переливаний реополиглюкина, препаратов гидроксиэтилкрахмала (инфукол, валекам, НЕS);
наличие волчаночного антикоагулянта;
мутация фактора IX;
дефекты при получении крови для исследования (гемолиз, передозировка цитрата натрия, забор крови из гепаринизированного катетера).
4.2. Протромбиновое время
Протромбиновое время (ПВ) – широко используемый скрининговый тест для оценки внешнего каскада свертывания плазмы. ПВ обычно используется для определения активности ф. VII, контроля за лечением непрямыми антикоагулянтами, при скрининге системы гемостаза, а также для количественного определения фибриногена в автоматических коагулометрах. Результаты определения протромбинового времени могут быть представлены в различной форме (табл.3).
Референсные значения ПВ: 9,2-12,2 с
УКОРОЧЕНИЕ ПВ:
активация внешнего механизма свертывания при различных видах внутрисосудистого свертывания крови;
последние недели беременности, прием пероральных контрацептивов;
лечение концентратами факторов протромбинового комплекса («Фейба», «НовоСевен» и др.).
УДЛИНЕНИЕ ПВ:
дефицит или аномалия факторов протромбинового комплекса (VII, X, V,II) в случаях приема антикоагулянтов непрямого действия (варфарин, синкумар, пелентан и др.);
болезни печени и желчевыводящей системы;
лечение нефракционированным гепарином (тест реагирует лишь на сравнительно высокие концентрации антикоагулянта, примерно от 0,5 МЕ/мл крови и выше);
ДВС-синдром (потребление факторов свертывания в переходную фазу и фазу гипокоагуляции);
на фоне переливаний реополиглюкина, препаратов гидроксиэтилкрахмала (инфукол, валекам, НЕS);
наличие в крови волчаночного антикоагулянта (возможно);
дефекты при получении крови для исследования (гемолиз, передозировка цитрата натрия, забор крови из гепаринизированного катетера).
Таблица 3. Формы выражения протромбинового времени
|
Показатель |
Расчёт |
Примечание |
Корреляция |
|
Протромбиновое время (ПВ), сек |
Время свёртывания плазмы после добавления тромбопластин-кальциевой смеси. |
Не позволяет проводить сравнительную оценку результатов в связи с применением тромбопластинов различного МИЧ (cм. МНО) |
|
|
Протромбиновый индекс(ПТИ), % |
ПВ нормальной контрольной плазмы (или среднее ПВ нормального диапазона)/ПВ плазмы пациента)х100 |
Результаты теста зависят от чувствительности используемого тромбопластина. |
Отрицательная – с протромбиновым отношением и МНО |
|
Протромбиновое отношение(ПО), % |
ПВ плазмы пациента/ПВ нормальной контрольной плазмы (или среднее ПВ нормального диапазона)х100 |
Результаты теста зависят от чувствительности используемого тромбопластина. |
Положительная – с МНО, отрицательная – с протромбиновым индексом |
|
Протромбин по Квику (%) |
Аналогично ПТИ, но расчёт производится в зависимости от концентрации факторов протромбинового комплекса |
Результаты теста зависят от чувствительности используемого тромбопластина. |
Отрицательная – с ПО и МНО, совпадает с ПТИ в области нормальных значений |
|
Протромбиновое время, выраженное через МНО - международное нормализованное отношение, латинская аббревиатура INR (International Normalized Ratio) (см. дальше по тексту). |
(ПВ плазмы пациента/ПВ нормальной контрольной плазмы (или среднее ПВ нормального диапазона)isi |
ISI (МИЧ) - (international sensitivity index - международный индекс чувствительности) – показатель чувствительности тромбопластина относительно международного стандарта (указывается в паспорте набора), что позволяет сравнивать между собой результаты, полученные с применением тромбопластина различной чувствительности. | |
Первые три выражения, хотя и представляются в виде цифр, но из-за отсутствия калибровки являются, по сути, качественными показателями с неопределенным масштабом. Кроме того, существенным недостатком определения протромбинового времени в секундах является низкая воспроизводимость из-за нестандартизированного тромбопластина. Поэтому нельзя сопоставлять результаты у одного пациента, полученные в разных лабораториях, на разных приборах или с тест - наборами разных серий. Выражение ПТИ в процентах не имеет смысловой нагрузки и путает врачей, так как между количеством факторов и измерением ПВ в секундах нет прямой пропорциональной зависимости. Протромбин по Квику и протромбиновое время, выраженное через МНО для ПВ являются взаимодополняющими. Протромбин по Квику (%) как и протромбиновый индекс, позволяет определять активность протромбинового комплекса плазмы пациента в сравнении с измеренным протромбиновым временем контрольной плазмы. Но при этом расчет проводится по кривой зависимости протромбинового времени от % содержания факторов протромбинового комплекса, построенной с использованием разных разведений контрольной плазмы. Такой способ представления результатов является более точным, особенно в области низких значений. Протромбиновый индекс и протромбин по Квику могут совпадать друг с другом в области нормальных значений. В зоне низких значений, рекомендованных для ведения больных, принимающих непрямые антикоагулянты, показатели этих тестов расходятся. Протромбиновый индекс 50-60% может соответствовать 30-40% протромбина по Квику. Расчет протромбина по Квику в настоящее время является общепринятым способом.
МНО
(Международное нормализованное
отношение), латинская аббревиатура INR
(International Normalized Ratio) - дополнительный
способ представления результатов
протромбинового теста, рекомендованный
для контроля терапии непрямыми
антикоагулянтами комитетом экспертов
ВОЗ, Международным комитетом по изучению
тромбозов и гемостаза и Международным
комитетом по стандартизации в гематологии.
МНО рассчитывается по формуле:
 где
ISI (International Sensitivity Index of thromboplastin), он же
МИЧ (международный индекс чувствительности)
- показатель чувствительности
тромбопластина, стандартизующий его
относительно международного стандарта.
МНО - математическая коррекция, при
помощи которой производится стандартизация
протромбинового времени, что позволяет
сравнивать результаты, полученные в
разных лабораториях. МНО и протромбин
по Квику коррелируют отрицательно -
снижение протромбина по Квику соответствует
повышению МНО.
где
ISI (International Sensitivity Index of thromboplastin), он же
МИЧ (международный индекс чувствительности)
- показатель чувствительности
тромбопластина, стандартизующий его
относительно международного стандарта.
МНО - математическая коррекция, при
помощи которой производится стандартизация
протромбинового времени, что позволяет
сравнивать результаты, полученные в
разных лабораториях. МНО и протромбин
по Квику коррелируют отрицательно -
снижение протромбина по Квику соответствует
повышению МНО.
Для контроля уровня антикоагулянтов ВОЗ разработаны следующие рекомендации:
|
Клиническое состояние |
Рекомендуемое МНО |
|
Профилактика первичного и повторного тромбоза глубоких вен и легочной тромбоэмболии |
2,5 (2,0-3,0) |
|
Предоперационная подготовка: хирургические вмешательства в области бедра |
2,0 (2,0-3,0) |
|
Все остальные хирургические вмешательства |
2,5 (1,5-2,5) |
|
Лечение тромбоза глубоких вен, легочной тромбоэмболии и профилактика повторного венозного тромбоза. |
3,0 (2,0-4,0) |
|
Профилактика артериальной тромбоэмболии, включая пациентов с искусственными клапанами |
3,5 (3,0-4,5) |
Рекомендуемые уровни гипокоагуляции при приеме варфарина:
высокий МНО от 2,5 до 3,0;
средний МНО от 2,0 до 3,0;
низкий МНО от 1,6 до 2,0.
4.3. Тромбиновое время
Тромбиновое время (ТВ) – определение тромбинового времени является третьим по значимости базисным скрининговым тестом. Тест характеризует конечный этап процесса свертывания – превращение фибриногена в фибрин под действием тромбина, на него влияет концентрация фибриногена в плазме и наличие продуктов деградации фибрина.
Референсные значения ТВ: 18-24 с
УКОРОЧЕНИЕ ТВ:
гиперфибриногенемия (фибриноген 6,0 г/л и выше);
начальная (гиперкоагуляционная) фаза острого и подострого ДВС-синдрома.
УДЛИНЕНИЕ ТВ:
гепаринотерапия обычным гепарином (тест реагирует на сравнительно низкие концентрации антикоагулянта, приблизительно от 0,05 МЕ/мл крови)
гипофибриногенемия (фибриноген ниже 1,0 г/л) в случаях развития острого ДВС-синдрома и при тромболитической терапии (стрептокиназа, актилизе и др.). В последнем случае конечный этап свертывания крови ингибируется продуктами деградации фибриногена и фибрина (фрагментами D и D-димеров)
влияние других ингибиторов полимеризации фибрин-мономера (парапротеины, миеломные белки и др.)
дефекты при получении крови для исследования (гемолиз, передозировка цитрата натрия, забор крови из гепаринизированного катетера)
4.4. Концентрация фибриногена в плазме
Количественное определение фибриногена по методу Клаусса является базисным тестом исследования гемостаза. Образование фибрина и его стабилизация представляют собой финальный этап формирования тромба, при котором растворимый фибриноген превращается в нерастворимый фибрин под действием тромбина и фактора XIII. Фибриноген – острофазный белок. Печень синтезирует 2–5 г фибриногена в день, время полувыведения фибриногена из крови составляет около 4 дней. Концентрация его может превышать 10 г/л при тяжелых бактериальных инфекциях, при травме и тромбозе. Повышение уровня фибриногена в острой фазе воспаления, как правило, имеет транзиторный характер. У курящих людей уровень фибриногена в плазме крови несколько выше, чем у некурящих. К значительному росту фибриногена приводят заболевания почек (пиелонефрит, гломерулонефрит, гемолитико-уремический синдром), коллагенозы (ревматоидный артрит, узелковый периартериит), пароксизмальная ночная гемоглобинурия, новообразования (рак легкого). При атеросклерозе наблюдается устойчивое увеличение уровня фибриногена, трудно корригируемое лекарственными препаратами. В результате риск сердечно-сосудистых заболеваний повышается с возрастанием исходного содержания фибриногена в интервале 3,0-4,5 г/л. Обнаружено, что повышение уровня фибриногена в плазме крови больных сердечно-сосудистыми заболеваниями предшествует развитию инфаркта миокарда и инсульта. Корреляция между уровнем фибриногена и развитием этих осложнений особенно четко прослеживается у пациентов молодого и среднего возраста. Определение уровня фибриногена – наиболее чувствительный тест для выявления бессимптомных стадий заболевания периферических артериальных сосудов.
Референсные значения фибриногена: 2,75- 3,65 г/л
СНИЖЕНИЕ КОНЦЕНТРАЦИИ ФИБРИНОГЕНА:
острый ДВС-синдром;
дисфибриногенемии.
ПОВЫШЕНИЕ КОНЦЕНТРАЦИИ ФИБРИНОГЕНА:
инфекционные, воспалительные и аутоиммунные процессы;
подострый и хронический ДВС-синдром;
нормально протекающая беременность.
5. Методы определения физиологических антикоагулянтов
5.1. Протеин С
Для определения протеина С разработано несколько методов: иммунохимический метод, метод с хромогенным субстратом и коагуляционный. Протеин С только в комплексе с протеином S инактивирует Vа и VIII, поэтому желательно параллельно исследовать и протеин С и протеин S. Определение протеина С с использованием хромогенного субстрата. Этот метод прост, специфичен, легко автоматизируется. Так как протеин С – витамин-К-зависимый гликопротеин, то при лечении непрямыми антикоагулянтами (антагонистами витамина К) появляются формы протеина С, которые не обладают антикоагулянтной активностью, но определяются методами с хромогенными субстратами (так называемые PIVKA-формы). Поэтому у больных, принимающих непрямые антикоагулянты, могут быть завышенными результаты определения протеина С хромогенными методами. Определение протеина С коагуляционным методом. Метод легко автоматизируется. Коагуляционный метод оценивает активность протеина С, фиксирующегося карбоксилированной частью на фосфолипидах. РIVKA -формы не функционируют в этой системе, и тест не дает ложных результатов у пациентов, принимающих непрямые антикоагулянты или имеющих дефицит витамина К. Исказить результаты теста могут такие факторы, как наличие мутации фактора V Лейден, присутствие в плазме волчаночного антикоагулянта, терапия гепарином. Для получения достоверных результатов очень важно качество используемой протеин-С-дефицитной плазмы. У некоторых пациентов, принимающих непрямые антикоагулянты, выявляются чрезвычайно низкие цифры содержания протеина С. Эти результаты могут быть связаны не только с истинным дефицитом протеина С, но и с развитием резистентности фактора Vа к активированному протеину С (АПС). Определение протеина С иммунохимическим методом. Иммунохимическое определение протеина С применяется реже, чем функциональные методы. Это объясняется тем, что данный способ выявляет только классическое снижение концентрации протеина С и не определяет его функциональную неполноценность. В некоторых случаях при лечении больных с дефицитом протеина С непрямыми антикоагулянтами у них могут возникнуть некрозы кожи в результате рикошетных тромбозов. Если антикоагулянты назначаются в высокой дозе без поддержки гепарином, то возникает состояние, при котором протеин С очень быстро снижается (в норме время его полужизни в системе циркуляции 4-6 ч). В то же время витамин-К-зависимые факторы свертывания, в том числе протромбин, остаются еще в пределах нормы (у них период полужизни 16-20 ч). Эта ситуация может усугубляться тем, что непрямые антикоагулянты начинают назначать в случае развития тромбозов или угрожающей ситуации по тромбообразованию, когда система прокоагулянтов активирована. Быстрое снижение протеина С провоцирует в этом случае массивное тромбообразование, проявляющееся вплоть до некрозов кожи. Активность протеина С следует определять до начала лечения антикоагулянтами непрямого действия и контролировать во время лечения.
Референсные значения протеина С: 94-124%
ПОВЫШЕНИЕ протеина С отмечается во время беременности.
СНИЖЕНИЕ протеина С:
врожденный (наследственный) дефицит или аномалии протеина С;
геморрагическая болезнь новорожденных;
заболевания печени с нарушением ее функции;
ДВС-синдром;
нефротический синдром;
синдром острой дыхательной недостаточности;
менингококковый сепсис;
гемодиализ;
лечении L-аспарагиназой;
лечении пероральными (непрямыми) антикоагулянтами (дефицит витамина К);
послеродовый и послеоперационный период.
5.2. Протеин S
Протеин S – витамин-К-зависимый белок, который является кофактором активированного протеина С. Это функция положена в основу всех известных коммерческих тест-систем определения протеина S. Описаны случаи как функционального, так и количественного дефицита протеина S. При проведении тестов важно помнить, что протеин S присутствует в плазме частично в свободном состоянии, частично в комплексе с С4-связывающим протеином (С4-СП), но активна только свободная форма протеина S. Определение протеина S коагуляционным методом. Для определения протеина S необходимо использовать тест-систему, содержащую очищенный активный протеин С, его субстрат – фактор Vа и дефицитную по протеину S плазму. Специфичность метода относительная, так как фактор V Лейден, высокий уровень ф.VIII и волчаночный антикоагулянт могут существенно влиять на результаты теста, а именно: с этими факторами часто проводят дифференциальную диагностику, определяя протеин S. Так как коагуляционный метод подвержен действию многих интерферирующих факторов и не стандартизирован, то предпочитают использовать иммунохимический метод. Определение протеина S иммунохимическим методом. Иммунохимический метод достаточно широко распространен. Наборы последних разработок позволяют определять «свободный протеин S» прямо без предварительной обработки. Недостатком иммунохимического метода является то, что он выявляет протеолитически неактивные формы протеина S, которые иногда появляются в плазме.
Референсные значения протеина S: 81-111%
УМЕНЬШЕНИЕ СОДЕРЖАНИЯ (активности) протеина S:
врожденный (наследственный) дефицит;
врожденное (наследственное) уменьшение свободной фракции протеина S;
заболевания печени с нарушением ее функции;
ДВС-синдром;
нефротический синдром;
системная красная волчанка;
лечение L-аспарагиназой;
лечение пероральными (непрямыми) антикоагулянтами;
прием эстрогенов (пероральных контрацептивов);
беременность, послеродовый период;
наличие аутоантител к протеину S.
5.3. Антитромбин III
Для определения активности антитромбина III (АТ) чаще всего используют метод с хромогенным субстратом. Антитромбин расщепляет субстрат, в результате чего образуется окрашенный продукт, количество которого зависит от исходной активности антитромбина III. Существуют также иммунохимические (турбидиметрия, нефелометрия) и коагуляционные методы. Тест может применяться для мониторинга лечения гепарином. Длительная гепаринотерапия может приводить к снижению активности АТ в плазме. Лечение высокими дозами гепарина, особенно нефракционированным гепарином, приводит к транзиторному снижению АТ по механизму потребления, особенно у больных с тяжелой патологией, при критических состояниях, при ДВС-синдроме, сепсисе, злокачественных опухолях. У новорожденных уровень АТ составляет около 50 % и достигает уровня взрослых к 6 мес. Небольшое снижение АТ наблюдается в середине менструального цикла, в пред- и послеродовом периоде, при токсикозах второй половины беременности, в послеоперационном периоде. Эти сдвиги более выражены у пациентов с группой крови А (II), а также у пожилых.
Референсные значения АТ: 86 - 116%
СНИЖЕНИЕ СОДЕРЖАНИЯ (активности) АТ:
врожденный (наследственный) дефицит или аномалии АТ (снижение активности или чувствительности к гепарину);
заболевания печени (опухоли, цирроз, алкогольный гепатит);
нефротический синдром (протеинурия свыше 5 г/л);
карцинома легких;
ДВС-синдром;
множественные травмы, тяжелые роды, поздние гестозы;
прием эстрогенов (пероральных контрацептивов), кортикостероидов;
лечение L-аспарагиназой.
УВЕЛИЧЕНИЕ СОДЕРЖАНИЯ (активности) АТ:
во время менструации;
острый вирусный гепатит, холестаз;
прием анаболических стероидов;
лечение пероральными (непрямыми) антикоагулянтами.
6.Тесты для исследования фибринолитической системы
Наиболее распространенные в клинической практике методы оценки состояния фибринолитической системы основаны на: 1) исследовании времени и степени лизиса (растворения) сгустков крови или эуглобулиновой фракции плазмы (общеоценочные пробы); 2) определении концентрации плазминогена, его активаторов и ингибиторов (ТАП; ПАИ-1; ?2-антиплазмин).
6.1. Время лизиса эуглобулиновых сгустков / ХIIа зависимый фибринолиз
Определение фибринолитической активности эуглобулиновой фракции плазмы крови является важнейшим базисным методом исследования системы фибринолиза, позволяющим оценить состояние внутреннего и внешнего механизмов образования плазминогена. Метод заключается в определении времени спонтанного лизиса сгустка, образующегося из эуглобулиновой фракции бестромбоцитной плазмы при добавлении к ней раствора хлорида кальция. Существуют и другие модификации метода эуглобулинового лизиса сгустка. Например, растворение сгустка может быть значительно ускорено предварительным введением в плазму каолина — мощного контактного активатора внутреннего механизма фибринолиза, связанного с активированием комплекса факторов: фактор XII-калликреин-кининоген (“ХIIа зависимый фибринолиз”). Метод оценки эуглобулинового лизиса требует исходного содержания в плазме фибриногена, так как при снижении фибриногена время лизиса укорачивается, что трактуется ошибочно как гиперфибринолиз. При гиперфибриногенемии время лизиса удлиняется. Поэтому при отклонениях содержания фибриногена в плазме, а также неполноценной полимеризации фибрина возможно получение ошибочных результатов. В связи с ориентировочным характером и недостаточной специфичностью в последнее время вместо теста спонтанного лизиса эуглобулинового сгустка начали использовать определение отдельных факторов фибринолиза, в первую очередь плазминогена.
Референсные значения: XIIа зависимый фибринолиз 4–10 мин
УКОРОЧЕНИЕ ВРЕМЕНИ ЛИЗИСА (активация фибринолиза):
уменьшение концентрации фибриногена – гипо- и дисфибриногенемия.
УВЕЛИЧЕНИЕ ВРЕМЕНИ ЛИЗИСА (угнетение фибринолиза):
гиперфибриногенемия.
6.2. Плазминоген и тканевой активатор плазминогена (ТАП)
Определение количества плазминогена основано на гидролизе хромогенного субстрата. Определение плазминогена используют для диагностики ДВС-синдрома и тромбофилий; выявления нарушений фибринолиза; контроля лечения фибринолитическими препаратами при тромбозах, тромбоэмболиях, инфарктах. Дефицит плазминогена крайне редкое событие, чаще встречается дефицит тканевого активатора плазминогена (ТАП). Дефицит ТАП является одним из потенциальных факторов риска тромбоза, хотя клинически это подтверждается не всегда. Тканевой активатор плазминогена (ТАП) освобождается в кровоток из эндотелиальных клеток сосудистой стенки при стрессовых воздействиях, в частности при манжеточной пробе (дозированном пережатии вен). Сначала определяют базовый уровень ТАП, потом на 10-15 минут на предплечье накладывают жгут или раздувают манжетку, вызывающую венозный стаз, затем берут вторую порцию крови, в которой повторно определяют ТАП. Сравнивают результаты обеих проб. ТАП обладает высокой амидазной активностью, позволяющей эффективно использовать для его определения метод хромогенных субстратов. Определение ТАП проводится у больных с тромбофилией как часть панели тестов на выявление причины тромбофилии, особенно при нагрузочных манжеточных пробах. Повышение ТАП после инфаркта миокарда рассматривается как неблагоприятный фактор. Нарушение освобождения ТАП после венозного стаза описано у больных с тромбозами и патологией почек.
Референсные значения плазминогена: 71-101%
УВЕЛИЧЕНИЕ СОДЕРЖАНИЯ ПЛАЗМИНОГЕНА И ЕГО АКТИВАТОРОВ:
панкреатит;
панкреонекроз;
метастазирующий рак предстательной железы, яичников;
метастазы меланомы;
операции на легких, предстательной, поджелудочной железе;
гиперкатехоламинемия (стресс, тиреотоксикоз, гипертонический криз, введение адреналина);
патология беременности;
терминальные и другие состояния, сопровождающиеся развитием ДВС-синдрома;
цирроз печени;
метастатическое поражение печени (снижение антиплазминовой и антиактиваторной функции).
ДЕФИЦИТ ПЛАЗМИНОГЕНА, ЧАЩЕ ДЕФИЦИТ ТКАНЕВОГО АКТИВАТОРА ПЛАЗМИНОГЕНА:
рецидивирующие венозные тромбозы;
системные васкулиты;
сепсис;
нефротический синдром.
7. Тесты активации свертывания крови
7.1. D-димеры
D-димеры – специфические продукты деградации фибрина, входящие в состав тромба. Они образуются в процессе лизиса сгустка крови под влиянием плазмина и некоторых неспецифических фибринолитиков. Концентрация D-димеров в сыворотке пропорциональна активности фибринолиза и количеству лизируемого фибрина. Этот тест позволяет судить об интенсивности процессов образования и разрушения фибриновых сгустков. Определение D-димеров проводится иммуноферментным методом с использованием моноклональных антител, иммунодиффузии, методом турбидиметрии, латекс-агглютинации. Во всех методах исследования используются моноклональные антитела к эпитопам на D-димере, которые образуются при расщеплении нерастворимого фибрина плазмином. Этих эпитопов нет на фибриногене и растворимых фибрин-мономерных комплексах (РФМК), поэтому D-димеры – показатель того, что в процессе фибринолиза расщепляется именно фибрин, а не фибриноген или фибрин-мономеры. Поскольку эти антитела не взаимодействуют с фибриногеном, исследования могут проводиться как в плазме, так и сыворотке. На определение D-димеров практически не оказывает влияние техника взятия крови, примесь тромбоцитов, не требуется использования ингибиторов для подавления других факторов.
Референсные значения D-димера: 33,5-727,5 нг/мл
ПОВЫШЕНИЕ уровня D-димеров в крови определяется при возникновении венозных тромбозов, атеротромбозе, тромбоэмболии легочной артерии, ДВС-синдроме, после операций, особенно при большом операционном поле и других состояниях с повышенным образованием фибрина. D-димеры достаточно долго циркулируют в крови, время их полувыведения составляет более 24 ч, повышение D-димеров может персистировать в течении нескольких недель после острого тромбоза. Уровень D-димеров повышен у больных с тромбозом глубоких вен бедра, с тромбоэмболией легочной артерии, он может повышаться после обширных хирургических вмешательств, травм, при онкологических заболеваниях. На содержание D-димеров влияют такие факторы, как величина тромба, время от начала клинических проявлений до назначения антикоагулянтной терапии, прием антикоагулянтов, на фоне которых уровень D-димеров постоянно снижается. Поэтому более важной для исключения диагноза тромбоза является отрицательная диагностическая значимость теста. Причем для разных методов определения Д-димеров отрицательная диагностическая значимость колеблется от 78 до 100%, она выше у более чувствительных методов, что характерно для ИФА-диагностики.
7.2.Растворимые фибрин-мономерные комплексы (РФМК)
При ряде форм патологии, характеризующихся активацией свертывания крови (ДВС, тромбозы, тромбофилии), происходит расширение пула фибриногена, в результате чего увеличевается количество растворимых фибрин-мономерных комплеков (РФМК). В последнее время стал активно использоваться ортофенантролиновый тест. С помощью ортофенантролинового теста возможно не только качественное, но и количественное определение РФМК. Гепаринотерапия с содержанием гепарина в плазме крови до концентрации 10 ед/мл не влияет на результаты теста.
Референсные значения: (РФМК по орто-фенантролиновому тесту) - до 4,0 мг%
ПОВЫШЕНИЕ:
активация внутрисосудистого свертывания крови (ДВС-синдром, тромбоз глубоких вен, эмболии легочной артерии);
возможно при лечении антикоагулянтами;
физический и психологический стрессы;
нормально протекающая беременность;
в период новорожденности .
8.Основные схемы обследования нарушений гемостаза
При ведении пациентов с подозрением на нарушения системы гемостаза, как ни в каком другом разделе медицины, важны результаты лабораторных исследований. Многие геморрагические заболевания имеют сходную клиническую картину, однако требуют разных терапевтических подходов. Только лабораторная диагностика позволяет установить точный диагноз и назначить современную адекватную терапию. Тромботические проявления, как правило, возникают при сочетании разных протромботических факторов, как врожденных, так и приобретенных. Лабораторная диагностика позволяет выявить эти факторы и провести не только лечение развившегося тромбоза, но и фоновых изменений и предупредить рецидивы тромбозов, контролируя терапию.
8.1.Определение причин кровоточивости
При наличии кровотечений провести диагностический поиск для большинства видов геморрагических диатезов может помочь приведенная ниже схема (табл. 4).
Таблица 4. Ориентировочная схема обследования при определении причин кровоточивости [3]
|
Основной метод |
Патология | |
|
Время кровотечения по Айви |
Более 10-12 мин | |
|
Количество тромбоцитов в крови |
Менее 80-100?109/л | |
|
Оценка агрегационной функции тромбоцитов с использованием таких индукторов, как АДФ, адреналин и коллаген |
Гипоагрегация | |
|
АПТВ |
Гипокоагуляция | |
|
ПТ |
Гипокоагуляция | |
|
Концентрация фибриногена |
Менее 1,0 г/л | |
|
Дополнительные методы, в случае наличия увеличения времени кровотечения и гипокоагуляции по АПТВ | ||
|
Фактор Виллебранда |
Менее 55% активности | |
|
Факторы VIII и IX |
Менее 40% активности | |
Вероятнее всего кровоточивость проявляется при сочетании или комбинации отдельных нарушений гемостаза, которые, усиливая друг друга, способствуют развитию геморрагического синдрома. Например, сама по себе гипофибриногенемия крайне редко вызывает кровотечение, но в сочетании с тромбоцитопенией/тромбоцитопатией либо дефицитом другого (других) факторов свертывания провоцирует развитие геморрагического синдрома. Такая ситуация может возникнуть при лечении тромболитиками. То же самое касается и оценки значимости тромбоцитопений различной степени выраженности. Для обеспечения тромбоцитарного гемостаза, как известно, достаточно содержания в крови 10-20?109/л этих клеток при условии, что они функционально активны и нет острой травмы (операции, родов и др.). Как правило, геморрагический синдром возникает при сочетании количественного и качественного дефектов кровяных пластинок. Это характерно, в частности, для ДВС-синдрома, когда при уровне кровяных пластинок ниже 100?109/л высока вероятность развития спонтанных кровотечений. По-видимому, в этом кроется и возможная причина разной выраженности и частоты возникновения гематом у больных гемофилией при дефиците факторов VIII или IX, в случае наличия или отсутствия сопутствующего снижения функции тромбоцитов, дисфибриногенемии и других факторов (например, при сочетании с мезенхимальной дисплазией), способствующих кровоточивости (табл 5).
Таблица 5. Классификация основных типов кровоточивости [3]
|
Тип кровоточивости |
Основные виды патологии |
|
Микроциркуляторный (петехиально-пятнистый, синячковый) |
Тромбоцитопении, тромбоцитопатии, болезнь Виллебранда |
|
Гематомный |
Гемофилии А и В |
|
Смешанный (микроциркуляторно-гематомный) |
ДВС-синдром (в стадии клинической манифестации), тяжелая степень болезни Виллебранда, передозировка прямых или непрямях антикоагулянтов, антиагрегантов, избыточная тромболитическая терапия |
|
Васкулитно-пурпурный |
Микротромбоваскулиты |
|
Ангиоматозный |
Телеангиэктазия, микроангиоматоз |
8.2. Диагностика болезни Виллебранда
Болезнь Виллебранда (БВ) относится к наиболее распространенным видам врожденных геморрагических диатезов (около 1% в популяции), передается аутосомно-доминантным путем, характеризуется количественной или качественной патологией фактора Виллебранда (ФВ) и клинически проявляется кровоизлияниями в кожу и слизистые оболочки. ФВ является белковым носителем для коагуляционного фактора VIII, защищает его от протеолиза. Для больных характерны носовые кровотечения, экхимозы, меноррагии, гематурия, чрезмерные кровотечения (в анамнезе) после незначительной травмы или хирургического вмешательства (при тонзилэктомии, удалении зуба). Заболевание выявляется в одной семье у лиц обоего пола, причем кровоточивость чаще обнаруживается у женщин. Перечисленные факторы дополняют диагностику и позволяют различить БВ и легкую форму гемофилии А, при которой эти симптомы отсутствуют. Подозрение на БВ может быть вызвано приведенной выше клинической картиной и удлинением показателей двух скрининговых («глобальных») тестов – время кровотечения и АПТВ. В последнем случае гипокоагуляция может быть обусловлена более или менее выраженным снижением активности фактора VIII в сочетании со снижением ФВ. Снижение уровня ФВ может быть одним из проявлений мезенхимальной геморрагической дисплазии.
Классификация и патогенез болезни Виллебранда
В соответствии с общепринятой классификацией (Sadler, 1994) БВ подразделяется на 3 типа, тип 2- на 4 подтипа:
1-й тип – наследственное заболевание с частичным дефицитом ФВ в крови и нормальным распределением мультимеров ФВ;
2-й тип – наследственная патология с качественным изменением ФВ. Второй тип разделяют на 4 подтипа:
подтип 2А - характеризуется снижением мультимеров ФВ большой и средней молекулярной массы;
подтип 2В – большие мультимеры ФВ снижены, увеличена аффинность к тромбоцитарному гликопротеину 1b и снижена аффинность к другим рецепторным гликопротеинам;
подтип 2М – характеризуется сниженной ристомицин-кофакторной активностью при нормальном количественном и качественном составе мультимеров ФВ;
подтип 2N – качественные варианты ФВ со значительным снижением аффинности к ф. VIII
3-й тип – практическое отсутствие ФВ в крови.
При градации степени тяжести БВ исходят из обычной клинической характеристики – частоты и степени выраженности кровотечений.
Таблица 6. Клинические особенности и диагностические признаки болезни Виллебранда [2]
|
Подтипы |
Частота встречаемости |
Клинические особенности |
Диагностика |
|
тип 1 |
1-30:1000; наиболее распространенный вариант БВ (>70% всех случаев). |
Легкие и умеренные признаки кровоточивости; аутосомно-доминантный тип наследования с неполной пенетрантностью (примерно 60%). |
Антиген ФВ, активность ФВ и ф. VIII снижена пропорционально (20-50%). Все фракции мультимеров представлены пропорционально. |
|
подтип 2А |
Приблизительно 10-15% всех клинически значимых случаев БВ. |
Легкие и умеренные признаки кровоточивости; чаще аутосомно-доминантное, но возможно и аутосомно-рецессивное наследование с более полной пенетрантностью, чем при 1 типе. |
Вариабельное снижение антигена ФВ, активности ФВ и ф. VIII, отсутствие высоко и среднемолекулярных мультимеров ФВ. |
|
подтип 2В |
Довольно редкий вариант (<5% всех клинически значимых случаев БВ). |
Легкие и умеренные признаки кровоточивости; чаще аутосомно-доминантное наследование с более полной пенетрантностью, чем при 1 типе. |
Возможно снижение антигена ФВ и ф. VIII; уменьшение высокомолекулярных мультимеров, положительная агрегация с низкими концентрациями ристомицина, легкая и умеренная тромбоцитопения. |
|
подтип 2М |
Редкий тип (единичные описания). |
Тяжесть геморрагических расстройств различна; аутосомно-доминантный тип наследования. |
Вариабельное снижение антигена ФВ и ф. VIII. Активность ФВ снижена относительно антигена, несмотря на присутствие высокомолекулярных мультимеров ФВ. |
|
подтип 2N |
Встречается редко. |
Разнообразные по тяжести геморрагические нарушения. Клинические нарушения сходны с нетяжелыми формами гемофилии А. Наследование аутосомное. |
Активность антигена ФВ и ФВ различны, часто нармальные. Диспропорционально снижен ф. VIII. Как правило, нормальное распределение мультимеров, снижение или отсутствие фактора VIII связывающего активность ФВ. |
|
тип 3 |
1-5:1 000 000 |
Тяжелые геморрагические нарушения; аутосомно-рецессивное наследование. |
Антиген ФВ, активность ФВ и ф. VIII значительно снижены или не определяются. |
Лабораторная диагностика БВ осложняется тем, что ФВ является белком острой фазы воспаления и его уровень возрастает во время стресса, физической нагрузки, беременности, при приеме гормональных контрацептивов, а также после хирургического вмешательства. Следовательно, такая диагностика наиболее информативна при отсутствии перечисленных воздействий и состояний. С другой стороны, помимо генетических форм могут наблюдаться симптоматические (вторичные) варианты БВ (синдром Виллебранда) в связи с нарушениями гормонального статуса у женщин, при стрессовых реакциях.
Алгоритм диагностики болезни Виллебранда [2]
Основные методы диагностики
время кровотечения
число тромбоцитов
АПТВ
активность фактора VIII
Подтверждающие тесты
ристоцетин-кофакторная активность ФВ с использованием агрегометра
антиген фактора Виллебранда (методом ИФА)
ристомицин-индуцированная агрегация тромбоцитов для диагностики подтипа 2В, при котором она повышена либо в норме
Специальные тесты
коллаген-связывающая активность ФВ
исследование мультимерного состава ФВ
определение тромбоцитарного ФВ
молекулярно-генетическая диагностика мутаций гена ФВ методом ПЦР
определение с использованием хромогенного субстрата
8.3.Распознавание врожденных и приобретенных тромбофилий
Проблема патологического тромбообразования – одна из важнейших терапевтических проблем в развитых странах. Ишемическая болезнь сердца, тромбоэмболия легочной артерии (ТЭЛА), тромбозы глубоких вен – патологические состояния, приводящие к тяжелой инвалидности и гибели человека. При лечении таких пациентов лабораторный контроль состояния гемостаза – важнейший фактор успеха. Как правило, тромбофилия – комбинированное состояние, возникающее вследствие действия нескольких патогенетических факторов. Основные патогенетические факторы тромбофилии:
Повреждение эндотелиальных клеток с обнажением тромбогенных субэндотелиальных структур.
Активация тромбоцитов циркулирующими агонистами либо вследствие взаимодействия тромбоцитов с субэндотелиальными структурами или фактором Виллебранда.
Активация свертывания крови.
Резистентность к антикоагулянтам или дефицит антикоагулянтов.
Снижение активности фибринолиза.
Реологические нарушения и стаз.
КЛАССИФИКАЦИЯ ТРОМБОФИЛИЙ (З.С. БАРКАГАН 1996, 2000 ГГ.)
I. Гемореологические формы тромбофилии
полицитемии и полиглобулии (идиопатические, гипоксические, дегидратационные, лейкемические)
нарушение формы, объема, деформируемости эритроцитов (в т.ч. гемоглобинопатии)
повышение вязкости плазмы (парапротеинемии, гиперфибриногенемии)
II. Тромбофилии тромбоцитарного происхождения
тромбоцитемии (первичные, симптоматические, в т.ч. неопластические)
гиперагрегационные формы (синдром "вязких" тромбоцитов первичный и при атеросклерозе, сахарном диабете, приёме гормональных контрацептивов)
повышение продукции или активности (мультимерности) фактора Виллебранда
III. Тромбофилии, обусловленные дефицитом или аномалиями физиологических антикоагулянтов
антитромбин III
кофактор гепарина II
протеин С
протеин S
увеличение уровня тромбомодулина в плазме
IV. Тромбофилии, связанные с дефицитом или аномалиями плазменных факторов свёртывания крови
аномалия фактора V (мутация Лейден) – АПС резистентность
симптоматические формы АПС резистентности (антифосфолипидный синдром и др.)
мутация протромбина G 202110А
дефицит фактора XII (прекалликреина)
дисфибриногенемии
V. Тромбофилии, связанные с повышением и/или гиперактивацией плазменных факторов свёртывания
повышение уровня фактора VIII свыше 150%
гиперактивация фактора VII часто при коронарной болезни сердца (КБС) у больных с тромбозами
высокий уровень фактора XI
высокий уровень фактора IX
гиперпродукция XIII фактора
VI. Тромбофилии, обусловленные нарушениями фибринолиза
дефицит или аномалии плазминогена
сниженное высвобождение тканевого активатора плазминогена (ТАП)
избыточное высвобождение ПАИ 1
избыток ?2-антиплазмина
Тромбофилии, аутоиммунного и инфекционно-иммунного генеза
АФС (первичный и вторичный)
при иммунных тромбоваскулитах
при системных иммунных заболеваниях (болезнь Бехчета)
при гипертрофической миокардиопатии
при инфекционно-иммунных заболеваниях (гемолитико-уремический сидром, затяжной бактериальный эндокардит)
Тромбофилии при обменных заболеваниях
при гипергомоцистеинемии
при сахарном диабете
при ожирении
при подагре
при гиперлипидемиях
Лекарственные формы тромбофилии
приём оральных контрацептивов
при длительной гепаринотерапии (тромбоцитопения, рикошетный тромбоз при дефиците антитромбина III
при лечении непрямыми антикоагулянтами кумаринами – варфарином (на фоне дефектов в системе протеина С)
при лечении тромболитиками (истощение плазминогена)
при лечении ?-аспарагиназой
Особые формы
при гемолитико-уремическом синдроме
при болезни Мошкович
онкотромбозы (синдром Труссо)
при микроангиопатической гемолитической анемии
Таблица 7. Краткосрочные и хронические факторы риска венозного тромбоэмболизма (ВТЭ)
|
Краткосрочные провоцирующие факторы |
Хронические риск факторы |
|
травмы ног хирургические вмешательства беременность рак, метастазы гипсование ног иммобилизация > 3 дней воздушные перелеты > 12 часов оральные контрацептивы гормональная заместительная терапия острые инфекции (сепсис) |
венозный тромбоэмболизм (ВТЭ) в анамнезе сердечная недостаточность обструктивная болезнь легких мембранозный нефрит нефротический синдром антифосфолипидный синдром тромбофилии, семейный анамнез миелопролиферативные заболевания пароксизмальная ночная гемоглобинурия легочная гипертензия гемиплегия, параплегия ожирение болезнь Крона венозная недостаточность полиглобулия эритроцитозы |
Важнейшие проявления и осложнения тромбофилий:
тромбоэмболии, ишемии и инфаркты;
невынашивание беременности и внутриутробная гибель плода;
злокачественная пурпура новорожденных;
кратное возрастание риска тромбоэмболий при беременности, гиперлипидемиях, хирургических вмешательствах;
высокий риск метастазирования злокачественных образований;
значительное повышение риска развития лекарственных тромбозов (контрацептивы, лечение цитостатиками);
увеличение риска тромбозов при всех видах полиглобулии и больших потерях жидкости.
Первичный лабораторный скрининг тромбофилий и отбор групп риска для профилактики тромбозов и ТЭЛА
Гемореология (полиглобулия и др.).
Количество тромбоцитов (более 500x109/л).
Активация и гиперагрегация тромбоцитов.
АПТВ, кроме гипокоагуляционных форм (гиперкоагуляция).
Концентрация фибриногена (свыше 5 г/л).
Уровень фактора VIII (свыше 150%).
Уровень РФМК в плазме (свыше 10 мг%).
Активность антитромбина III (менее 70%).
Уровень протеина С и S (нормализованное отношение менее 0,8 (в парус-тесте, Барнаул)).
Волчаночный антикоагулянт (наличие в плазме).
Уровень гомоцистеина в крови (по ИФА выше 11 мкг/мл).
Нарушение фибринолиза (снижение плазминогена и его активаторов, повышение ингибиторов ПАИ-1; ?2-антиплазмина).
ПЦР – диагностика (наличие мутации Лейден, протромбина или тетрагидрофолатредуктазы).
8.4. Диагностика антифосфолипидного синдрома и выявление аутоантител, обладающих свойствами волчаночного антикоагулянта (ВА)
Антифосфолипидный синдром (АФС) известен как приобретенный аутоиммунный процесс, в основе которого лежит образование в организме в высоком титре бимодальных аутоантител, взаимодействующих с отрицательно заряженными мембранными фосфолипидами и связанными с ними гликопротеинами. Среди мембранных фосфолипидов основными мишенями антифосфолипидных антител (АФА) являются несущие отрицательный заряд кардиолипин, фосфатидилсерин, фосфатидилэтаноламин и фосфатидиловая кислота, а из белковых компонентов – ?2-гликопротеин-1, аннексин V и протромбин (фактор II). Большинство длительно действующих АФА принадлежат к классам IgG и IgM. Они блокируют фосфолипидно-белковые комплексы как свободных фосфолипидных микровезикул плазмы крови, так и лабилизированных клеточных мембран эндотелия, тромбоцитов и других клеток. Это проявляется, с одной стороны, снижением тромборезистентности эндотелия и активацией тромбоцитарного гемостаза, а с другой – дисбалансом в системе коагуляционного гемостаза. Последний характеризуется развитием тромбофилического статуса in vivo, при котором активация факторов Vа, Xа и протромбина сочетается с депрессией противосвертывающих механизмов, в частности в системах протеина С (вариант вторичной резистентности фактора Vа к АПС), тромбомодулина и фибринолиза. Эти нарушения проявляются in vitro развитием гипокоагуляции в так называемых фосфолипидзависимых тестах. Эта же гипокоагуляция выявляется с помощью комплекса специальных лабораторных методик и обозначается как эффекты АФА,обладающих свойствами ВА.
КЛАССИФИКАЦИЯ АФС [3]
Первичный АФС:
политромботический синдром, нарушение мозгового кровообращения, особенно у лиц молодого возраста;
привычное невынашивание беременности и внутриутробная гибель плода (упорно повторяющиеся выкидыши при отсутствии акушерско-гинекологической патологии);
аллергия к лекарственным препаратам (хинидин, гидролазин, фенотиазин, кокаин, прокаинамид).
Вторичный АФС:
на фоне аутоиммунных заболеваний (системная красная волчанка (СКВ), узелковый периартериит, ревматоидный артрит, системная склеродермия, иммунный тиреоидит);
на фоне злокачественных новообразований (солидные опухоли, тимома, карциномы, гематологические опухоли);
на фоне инфекционных и инфекционно-иммунных заболеваний (болезнь Лайма, бронхиальная астма, ВИЧ-инфекция, стафилококковая, стрептококковая инфекция);
в связи с другими причинами (терминальная стадия почечной и печеночной недостаточности);
Другие варианты АФС:
катастрофический;
гипопротромбинемический;
микроангиопатический;
на фоне ДВС-синдрома.
Клинико-лабораторные признаки АФС
Для постановки диагноза «Антифосфолипидный синдром» требуется наличие у пациента как минимум одного клинического и одного лабораторного признаков.
I.Клинические признаки АФС
При первичном АФС:
Немотивированные рецидивирующие тромбозы сосудов (чаще вен) у 70-75% больных, в том числе тромбоэмболия легочной артерии (ТЭЛА).
Расстройства мозгового кровообращения (тромботический инсульт, мигренеподобные немотивированные головные боли, нарушение зрения, памяти, парезы, эпи-синдром).
Фетоплацентарная недостаточность с невынашиванием беременности (привычные, чаще поздние выкидыши, внутриутробная гибель плода).
Кровоточивость микроциркуляторного типа (20-25% больных).
Склонность к развитию ДВС-синдрома.
Тромбоцитопения с повышенной агрегацией тромбоцитов.
Гипокоагуляция крови, особенно в фосфолипид-чувствительных тестах, выполненных на бедной тромбоцитами плазме.
Ложноположительная реакция Вассермана.
Наличие вирусной инфекции (вирус гепатита В, цитомегаловирусы, вирус Эпштейн-Барр).
Наличие сопутствующих признаков других иммунных процессов (артралгии, аллергии к лекарствам, укусам насекомых, пищевым продуктам), не позволяющих отнести выявленную совокупность симптомов к четко очерченной нозологической форме.
При вторичном АФС:
Развитие АФС на фоне уже имеющегося иммунного (СКВ, узелковый периартериит, геморрагический васкулит и др.; примерно в 25-35% случаев при СКВ) или опухолевого (лимфосаркома, лимфогранулематоз, миеломная болезнь) процесса.
Поливалентная гаптеновая лекарственная аллергия.
II. Лабораторные критерии АФС
Антитела к ?2-гликопротеину-1 (IgG и/или IgM изотипа) в сыворотке или плазме, обнаруживающиеся в двух и более случаях с интервалом повторного анализа не менее 6 недель.
Волчаночный антикоагулянт (ВА) обнаруживается в плазме в 2-х или более образцах с промежутком не менее 6 нед (согласно руководству Международного общества по тромбозу и гемостазу).
Определение антикардиолипиновых антител (IgG и/или IgM изотипа) в сыворотке или плазме крови в двух или более случаях с интервалом повторного анализа не менее 6 недель.
Специфичность диагностики по антителам различна: антитела против ?2-гликопротеина-1 демонстрируют большую специфичность, чемантитела к кардиолипину или другим фосфолипидам. Основной недостаток ИФА заключается в том, что нельзя разграничить антитела, обладающие или не обладающие свойствами волчаночного антикоагулянта. Поэтому данный вид диагностики является вспомогательным и не может заменить распознавание ВА на базе коагуляционных тестов. Однако в 3-10% случаев у больных с АФС обнаружение антител к ?2-гликопротеину-1 является единственным лабораторным проявлением.
Этапы выявления волчаночных антикоагулянтов
Скрининговые фосфолипидчувствительные тесты
АПТВ
тест с разведенным ядом гадюки Рассела (со скрининговым реагентом)
тест ингибирования разведенного тканевого тромбопластина
каолиновое время свертывания бедной тромбоцитами плазмы (БТП)
Тест на смешивание плазмы больного с контрольной нормальной плазмой для исключения дефицита факторов свертывания (при наличии гипокоагуляции)
АПТВ
Тесты коррекции имеющейся гипокоагуляции фосфолипидами
Подтверждающие тесты на ВА
тест с разведенным ядом гадюки Рассела (со специальным реагентом)
тест нейтрализации тромбоцитами
8.5.Актуальные вопросы диагностики острого и подострого ДВС-синдрома
Отдельная задача ставится клиницистами перед лабораторией при распознавании и контроле за лечением такого спутника многих критических состояний, как ДВС-синдром. ДВС-синдром – неспецифический общепатологический процесс, связанный с поступлением в кровоток активаторов свертывания крови и агрегации тромбоцитов, диссеминированным микросвертыванием крови, активацией и истощением плазменных протеолитических систем, потреблением физиологических антикоагулянтов и факторов свертывания крови, образованием в зоне микроциркуляции микросгустков и агрегатов клеток крови, следствием чего является развитие блокады микроциркуляции в органах-мишенях, гипоксии, дистрофии и глубокой дисфункции этих органов. Данные нарушения сопровождаются интоксикацией организма продуктами тканевого распада, вторичной эндогенной бактериемией и развитием тяжелого тромбо-геморрагического синдрома.
Основные звенья патогенеза ДВС-синдрома
начальная активация гемокоагуляционного каскада и тромбоцитов эндогенными факторами: тканевым тромбопластином, лейкоцитарными протеазами, продуктами распада тканей, опухолевыми прокоагулянтами;
персистирующая тромбинемия с повышением уровня ее маркеров в крови (РФМК и D-димеров);
истощение системы физиологических антикоагулянтов со значительным снижением содержания в плазме антитромбина III, протеина С, плазминогена и повышением уровня тромбомодулина в плазме крови;
системное поражение сосудистого эндотелия и снижение его антитромботического потенциала;
образование микросгустков крови и блокада микроциркуляции в органах-мишенях (мозг, надпочечники, почки, печень, желудок и кишечник (субсиндром полиорганной недостаточности) с развитием дистрофических и деструктивных нарушений в них).
активация фибринолиза в зоне блокады микроциркуляции и истощение его резервов в общей циркуляции;
потребление факторов гемокоагуляции и тромбоцитопения (и - патия) потребления, приводящие к системной кровоточивости и терминальной гипокоагуляции вплоть до полной несвертываемости крови (геморрагическая фаза синдрома);
нарушение барьерной функции слизистой оболочки желудка и кишечника с трансформацией асептического ДВС-синдрома в септический;
вторичная тяжелая эндогенная интоксикация.
Менее четко очерчен хронический ДВС-синдром, при котором длительная волнообразно текущая фибринация сопровождается персистирующей тромбинемией, выраженной дисфункцией органов-мишеней при минимальной и зачастую моноорганной геморрагической симптоматике, но с одновременным возникновением тромбозов магистральных вен.
Этиологические формы острого и подострого ДВС-синдрома
Инфекционно-септические:
бактериальные;
вирусные;
токсически-шоковый (в том числе при абортах).
Травматические и при деструкциях тканей:
ожоговый;
синдром длительного сдавления;
массивные травмы;
при некрозах тканей и органов (острая токсическая дистрофия печени, некротический панкреатит, острый инфаркт миокарда и др.);
при остром внутрисосудистом гемолизе, в том числе при переливаниях несовместимой крови;
при травматичных операциях;
при массивных гемотрансфузиях;
при гемобластозах, прежде всего при остром промиелоцитарном лейкозе;
при острой лучевой болезни.
Акушерские и гинекологические:
при эмболии околоплодными водами (особенно инфицированными);
при ранней отслойке и предлежании плаценты;
при атонии и массаже матки;
при внутриутробной гибели плода и его ретенции;
при эклампсии.
Шоковые (при всех терминальных состояниях).
В процессе интенсивной химиотерапии.
При трансплантации органов.
Причинами хронического (затяжного) ДВС-синдрома чаще всего являются следующие виды патологии:
хрониосепсис, включая затяжной септический эндокадит;
хронические иммунные и иммунокомплексные болезни;
хронические вирусные заболевания (гепатит, ВИЧ и др.);
опухолевые процессы (рак, лимфомы, лейкозы и др.).
Диагностика ДВС-синдрома строится прежде всего на ситуационной основе, с выявлением всех возможных условий и видов патологии, в том числе и критических состояний, при которых развитие этого синдрома является закономерным, с учетом проявлений его клинической манифестации и данных лабораторного обследования больных. Все эти три подхода имеют самостоятельное значение и дополняют друг друга. Так, например, острый ДВС-синдром нередко дебютирует с профузного кровотечения, сопровождает шок любой этиологии, быстро приводит к полиорганной недостаточности, и эти ситуации нуждаются не в лабораторном подтверждении диагноза и неоправданной потере времени, а в скорейшей патогенетической терапии. Роль лабораторной диагностики при этом представляется важной для уточнения тяжести и этапа развития данного синдрома по степени потребления основных компонентов системы гемостаза (тромбоцитов, фибриногена, физиологических антикоагулянтов (антитромбина III, протеина С)), а так же в подборе и оценке эффективности проводимой терапии. Тем не менее в отдельных случаях, например при подостром течении ДВС-синдрома, роль лабораторной диагностики по известным критериям (табл. 6) становится определяющей, особенно в тех случаях, когда его клинические признаки (полиорганная недостаточность, кровоточивость) еще мало выражены или запаздывают.
Таблица 8. Ориентировочная схема обследования для диагностики острого и подострого ДВС-синдрома [3]
|
Метод |
Патология |
|
Количество тромбоцитов в крови |
Чаще тромбоцитопения |
|
АПТВ |
Фазовые изменения |
|
Протромбиновое время |
Фазовые изменения |
|
Тромбиновое время |
Фазовые изменения |
|
Уровень растворимого фибрина (или РФМК) и D-димера в плазме |
Повышение |
|
Концентрация фибриногена |
Широкий диапазон значений |
|
Активность антитромбина III |
Менее 70 % |
Преобладающее значение в лабораторной диагностике ДВС-синдрома принадлежит не выявлению гипер- или гипокоагуляционного сдвига и гипофибриногенемии (которая характерна лишь для молниеносных форм патологии и терминальной фазы глубокой несвертываемости крови), а выявлению тромбоцитопении, высокого уровня маркеров тромбинемии (растворимого фибрина и D-димера) и, что важно, потребления физиологических антикоагулянтов, степень снижения которых, наряду с глубиной тромбоцитопении и выраженностью клинических проявлений, отражает тяжесть ДВС-синдрома. Следует избегать перегруженного списка методов, иногда рекомендуемых для экспресс -диагностики ДВС-синдрома, поскольку это задерживает ответ, снижает информативность и своевременность реальной диагностики.
При распознавании и мониторировании острых и подострых ДВС-синдромов всегда необходимо учитывать возможное влияние на результаты исследований гепаринемии (при анализе крови, полученной через гепаринизированный катетер или при терапии гепарином), гемодилюции, наблюдающейся при массивной инфузионной терапии, гиперцитратемии и ряда плазмозаменителей, особенно реополиглюкина.
10.Гипокоагуляционно-геморрагические состояния
Геморрагический синдром (склонность к появлению кожных геморрагий и кровоточивости слизистых оболочек) развивается вследствие изменений в одном или нескольких звеньях гемостаза - при поражениях сосудистой стенки, уменьшении количества тромбоцитов или расстройстве их функций, нарушениях свёртывания крови. Из наследственных нарушений гемостаза в терапевтической практике наиболее часто наблюдают тромбоцитопатии, гемофилию А и B, болезнь фон Виллебранда, а из сосудистых форм - телеангиэктазии. Наиболее частые приобретённые формы геморрагического синдрома - вторичные тромбоцитопении и тромбоцитопатии, ДВС-синдром, дефицит факторов протромбинового комплекса и геморрагический васкулит. Другие формы наблюдают редко. Следует учитывать, что в последние годы нарушения гемостаза всё чаще связаны с приёмом ЛС (особенно антиагрегантов и антикоагулянтов).
Типы кровоточивости
В клинической практике выделяют пять типов кровоточивости.
• Гематомный тип характерен для гемофилии А и В, характеризуется возникновением болезненных напряжённых кровоизлияний в мягкие ткани, а также в суставы с постепенным нарушением функции опорно-двигательного аппарата.
• Петехиально-пятнистый (синячковый) тип возникает при тромбоцитопениях, тромбоцитопатиях, некоторых нарушениях свёртывающей системы, гипо- и дисфибриногенемиях, дефиците факторов протромбинового комплекса (VII, X, V и II).
• Смешанный синячково-гематомный тип развивается при выраженном дефиците факторов протромбинового комплекса и фактора XIII, болезни фон Виллебранда, ДВС-синдроме, передозировке антикоагулянтов и тромболитических препаратов, появлении в крови иммунных ингибиторов факторов VIII и IX. Проявляется сочетанием петехиально-пятнистых кожных геморрагий с отдельными большими гематомами в забрюшинном пространстве, стенке кишечника. В отличие от гематомного типа кровоточивости, кровоизлияния в суставы возникают крайне редко.
• Васкулито-пурпурный тип наблюдают при инфекционных и иммунных васкулитах (см. главу 49 "Системные васкулиты", легко трансформируется в ДВС-синдром и характеризуется геморрагиями в виде сыпи или эритемы, возможным присоединением нефрита и кишечных кровотечений.
• Ангиоматозный тип развивается в зонах телеангиэктазий, ангиом, артерио-венозных шунтов и характеризуется упорными локальными геморрагиями, связанными с зонами сосудистой патологии.
С определённой долей вероятности предположить патологию сосудисто-тромбоцитарного или коагуляционного звеньев гемостаза можно по особенностям геморрагических проявлений (табл. 56-1).
Таблица 56-1. Признаки нарушения сосудисто-тромбоцитарного и коагуляционного гемостаза
|
Клинические признаки |
Нарушения коагуляционногогемостаза |
Нарушения сосудисто-тромбоцитарного гемостаза |
|
Петехии |
Редко |
Характерны |
|
Расслаивающие гематомы |
Характерны |
Редко |
|
Поверхностные экхимозы |
Чаще большие одиночные |
Обычно небольшие множественные |
|
Гемартрозы |
Характерны |
Редко |
|
Отсроченная кровоточивость |
Характерна |
Редко |
|
Кровотечения из порезов и царапин |
Минимальные |
Длительные, часто интенсивные |
|
Преобладающий пол |
Мужской (80-90%) |
Женский (незначительно) |
|
Положительный семейный анамнез |
Часто |
Крайне редко |