

975741
.pdfО^обливості^бміну речовин
в мітохондріях клітин і зазнають обміну в циклі Кноопа-Лінена, суть якого полягає в тому, що при кожному оберті циклу утворюється одна молекула ацетил-коензиму А, і ланцюг жирної кислоти скорочується на два вуглеводних атоми.
Однак, незважаючи на великий приріст енергії при розщепленні жирів, організм віддає перевагу використанню як джерела енергії вуглеводів, оскільки можливості аутокаталітичної регуляції приросту енергії в циклі Кребса зі сторони шляхів обміну вуглеводів більші, ніж при обміні жирів.
При катаболізмі жирних кислот відбувається утворення проміжних продуктів — кетонових тіл (Р-оксимасляна кислота, ацетооцтова кислота і ацетон). їх кількість має певне значення, оскільки безпосередньо впливає на кислотно-лужний стан. Вуглеводи їжі і частина амінокислот мають також кетогенні властивості. Спрощено кетогенність дієти можна подати у вигляді такої формули: (жири + 40% білків)/(вуглеводи + 60%> білків). Якщо це співвідношення перевищує 2, то дієта має кетогенні властивості.
Слід мати на увазі, що незалежно від виду їжі існують вікові особливості, що визначають схильність до кетозу. Діти віком від 2 до 10 років особливо до нього схильні, а новонароджені і діти першого року життя, навпаки більш стійкі. Можливо, фізіологічне «дозрівання» активності ензимів, що беруть участь в кетогенезі, відбувається повільно. Утворення кетонових тіл здійснюється переважно в печінці. Однією з причин підвищеного утворення кетонових тіл може бути зниження концентрації глюкози та відносно високий рівень НЕЖК . При цьому зменшення окислення кетонових тіл в тканинах та порушення шляхів їх виділення з організму можуть призводити до збільшення їх концентрації в крові. Крім того, в розвитку кетозу суттєву роль відіграють порушенння амінокислотного обміну. Доведно, що лейцин, ізолейцин, тирозин, фенілаланін є кетогенними, а триптофан, лізин і глутамат — антикетогенними.
Таким чином, утворення, метаболізм та виведення кетонових тіл залежать від стану процесів обміну як ліпідів, так вуглеводів і білків.
На відміну від дорослих, у дітей мають місце вікові особливості ліпідограми крові (табл. 47). Як видно з табл. 47, вміст загальних ліпідів у крові підвищується з віком: тільки протягом першого року життя він збільшується майже в 3 рази. У новонароджених відносно високий вміст (в процентах від загальної кількості жиру) нейтральних ліпідів. На першому році життя значно зростає вміст лецитину при відносній стабільності кефаліну і лізолецитину.
Відомо, що а- ліпопротеїни містять 2/3 фосфоліпідів і близько 1/4 холестерину плазми крові, Р-ліпопротеїни — 3/4 холестерину і 1/3 фосфоліпідів. У новонароджених кількість (Х-ліпопротеїнів значно більша, тоді як Р- ліпопротеїнів небагато. Тільки в 4 міс. співвідношення а- і Р-фракцій ліпопротеїнів наближається до нормальних для дорослого величин (а-ліпо- Протеїнів 20-25%, Р-ліпопртеїнів — 75-80%). Це має певне значення для транспорту фракцій жиру.
231

Розділ 9
|
|
|
|
|
Таблиця 47 |
Вікові особливості вмісту жиру і його фракцій у дітей |
|||||
|
|
|
|
|
|
Показник |
|
|
Вік дитини |
|
|
|
1 год |
24 год |
6-10 днів |
1-2 міс |
2-14 років |
Загальні ліпіди, г/л |
2 |
2,21 |
4,7 |
5 |
6,2 |
|
|
|
|
|
(4,5-7,0) |
Тригліцериди, ммоль/л |
0,2 |
0,2 |
0,6 |
0,39 |
0,93 |
Холестерин, ммоль/л |
1,3 |
- |
2,6 |
3,38 |
5,12 |
|
|
|
|
|
(3,74-6,50) |
НЕЖК, ммоль/л |
2,2 |
2 |
1,2 |
0,8 |
0,45 |
Фосфоліпіди, ммоль/л |
0,65 |
0,65 |
1,04 |
1,6 |
2,26 |
|
|
|
|
|
(1,78-2,6) |
Лецитин, г/л |
0,54 |
- |
0,8 |
1,25 |
1,5 |
В крові новонароджених значно нижчий вміст ліпопртеїнів, ніж у дорослих, причому повністю відсутні хіломікрони, а вміст пребета-ліпопротеїнів (ліпопротеїни дуже низької щільності) різко знижений. Основним класом ліпопротеїнів у новонароджених є а-ліпопротеїни (ліпопротеїни високої щільності). Розподіл окремих класів ліпопротеїнів крові такий: пребеталіпопротеїни — 3-15%, (3-ліпопротеїни (ліпопротеїни низької щільності) — 3541%, а-ліпопротеїни — 50-56%. Сумарний вміст (3-ліпопротеїнів і пребеталіпопротеїнів становить 1,7 г/л, тобто в 5 разів нижчий, ніж в крові матері.
Пребета-ліпопротеїни у новонароджених характеризуються більшим вмістом білка та меншим — тригліцеридів, ніж пребета-ліпопротеїни у дорослих. На відміну від цього в Р-ліпопротеїнах у новонароджених відмічається більший вміст тригліцеридів, ніж у дорослих, більший вміст білка та неестерифікованого холестерину.
Склад а-ліпопротеїнів новонароджених характеризується більшим вмістом фосфоліпідів, неестерифікованого холестерину та меншим вмістом білка і тригліцеридів, порівняно зі складом материнських а-ліпопротеїнів. Частка холестерину в них в 2 рази більша, ніж у дорослих.
З віком рівень а - ліпопротеїнів знижується, а Р - ліпопротеїнів — підвищується. Однак у дітей від 2 до 14 років вміст Р-ліпопротеїнів не змінюється.
Порушення обміну жиру може відбуватися на різних етапах метаболізму. У зв'язку з цим слід розрізняти спадкові захворювання, обумовлені генетичними дефектами, та захворювання, які виникають в процесі життя.
Порушення перетравлення та всмоктування жирів може бути при закупорці жовчних протоків різної етіології, при гіперсекреції соляної кислоти, яка інактивує панкреатичну ліпазу (синдром Золлінгера-Еллісона), при патології слизової оболонки травного каналу (коліт, гастрит та ін.). В результаті цих станів порушується розщеплення і всмоктування харчових ліпідів та жиророзчинних вітамінів, а в фекаліях з'являється жир — стеаторея.
232

Особливості обміну речовин
Хоч і рідко, але спостерігається описаний Шелдоном синдром мальабсорбції жиру, обумовлений відсутністю панкреатичної ліпази. Клінічно це проявляється целіакоподібним синдромом зі значною стеатореєю. Внаслідок цього маса тіла хворих збільшується повільно. Виявляється і зміна еритроцитів в результаті порушення структури їх оболонки і строми. Схожий стан виникає після оперативних втручань на кишках, при яких резекуються значні їх відділи.
Порушення процесу переходу жира із крові в тканини мають місце при деяких генетично обумовлених захворюваннях, таких як абеталіпопротеїнемія, гіпоальфаліпопротеїнемія, гіперхіломікронемія, сімейна гіперхолестеринемія, гіпербеталіпопротеїнемія та ін.
Із захворювань, в основі яких лежить порушення транспорту жиру, відома абеталіпопротеїнемія — відсутність (3-ліпопротеїнів низької щільності. Суть дефекту полягає в неспроможності організму синтезувати ліпопротеїни низької щільності і в ймовірному порушенні засвоєння харчових жирів (утворення хіломікронів). Клінічна картина цього захворювання схожа на целіакію (діарея, стеаторея, гіпотрофія тощо). В крові вміст жиру низький (сироватка прозора). Відмічається зниження рівня холестерину, фосфоліпідів і тригліцеридів в плазмі крові. В міру прогресування захворювання виникає пігментна дегенерація сітківки, прогресуюча демієлінізація мозочка та бокових стовпів спинного мозку, що викликає мозочкову атаксію.
Однак частіше спостерігаються різні гіперліпопротеїнемії. Згідно з класифікацією ВООЗ, розрізняють п'ять типів: І — гіперхіломікронемія; II — гіпербета-ліпопротеїнемія; III — гіпербета-гіперпребета-ліпопротеїнемія; IV
— гіперпребета-ліпопротеїнемія; V — гіперпребета-ліпопротеїнемія і хіломікронемія.
Залежно від зміни сироватки і вмісту фракцій жиру їх можна розрізняти за прозорістю.
В основі І типу лежить дефіцит ліпопротеїнліпази, в сироватці крові міститься велика кількість хіломікронів, внаслідок чого вона каламутна. Нерідко виявляються ксантоми. Хворі часто страждають на панкретит, який супроводжується приступами гострого болю в животі; виявляється також і ретинопатія.
II тип характеризується підвищенням в крові рівня (ї-ліпопротеїнів низької Щільності з різким збільшенням вмісту холестерину і нормальним або дещо підвищеним рівнем тригліцеридів. Клінічно нерідко виявляються ксантоми на Долонях, сідницях, периорбітально та ін. Рано розвивається атеросклероз. Деякі автори виділяють два підтипи: Па і Пб.
ПІ тип — підвищення вмісту так званих флотуючих (3-ліпопротеїнів, високий вміст холестерину, помірне підвищення рівня тригліцеридів. Нерідко Уявляються ксантоми.
IV тип — підвищення вмісту пребета-ліпопротеїнів зі збільшенням числа тРигліцеридів, нормальним або дещо підвищеним рівнем холестерину;
Хіломікронемія |
відсутня. |
30-747 |
233 |
Розділ 9
V типу властиве підвищення вмісту ліпопротеїнів низької щільності зі зменшенням очищення плазми від харчових жирів. Захворювання клінічно проявляється болем у животі, хронічним рецидивуючим панкреатитом, гепатомегалією. Цей тип у дітей зустрічається рідко.
Гіперліпопротеїнемія — часто генетично обусловлене захворювання. Однак цей стан розвивається вторинно при різних захворюваннях (червоний вовчак, панкреатит, цукровий діабет, гіпотиреоз, нефрити, холестатичні жовтяниці та ін.). Він викликає раннє ураження судин — атеросклероз, формування ішемічної хвороби серця, небезпеку розвитку крововиливів в мозок.
Поряд з цим відомі і внутрішньоклітинні ліпоїдози, серед яких у дітей найчастіше зустрічаються хвороби Німана-Піка і Гоше. При хворобі НіманаПіка спостерігається відкладання в клітинах системи макрофагів, кістковому мозку сфінгомієліну, а при хворобі Гоше — гексозоцереброзидів. Одним з основних клінічних проявів цих хвороб є спленомегалія. У дітей першого року життя спостерігається хвороба Німана-Піка, а у старших одного року — хвороба Гоше.
9.4. Обмін вуглеводів
Вуглеводний обмін включає розщеплення вуглеводів в травному каналі до моносахарів, синтез і розпад глікогену в тканинах, анаеробне і аеробне розщеплення глюкози та глюконеогенез.
Вуглеводний обмін у дітей характеризується значно більшою інтенсивністю, ніж у дорослих. Для них також властива висока лабільність обміну вуглеводів, виражена тим більше, чим менша дитина. Діти, особливо раннього віку, не переносять безвуглеводної їжі. Так, у дітей раннього віку після двох днів безвуглеводного харчування цукор крові натще може знизитись на 1,3-1,6 ммоль/л, тоді як у дорослих за тих же умов лише на 0,6-1,2 ммоль/л.
Вуглеводи виконують в організмі людини важливі і різноманітні функції. Вони є основним джерелом енергії, у вигляді мукополісахаридів входять до складу сполучної тканини, а у вигляді складних сполук (глікопротеїди, ліпополісахариди) є структурними елементами клітин, а також складовими частинами деяких біологічно активних речовин (ферменти, гормони, імунні тіла тощо). Частка вуглеводів у раціоні харчування дітей значною мірою залежить від віку. У дітей першого року життя вміст вуглеводів, які забезпечують потребу в калоріях, становить 40%. Після року він збільшується до 60%. У перші місяці життя потреба у вуглеводах покривається за рахунок молочного цукру (лактози), що входить до складу жіночого молока. При штучному вигодовуванні молочними сумішами дитина також отримує сахарозу або мальтозу. Після введення прикорму дитина починає одержувати полісахариди (крохмаль, частково глікоген), які в основному покривають потреби організму у вуглеводах.
234
Особливості обміну речовин
Т а к ий характер харчування дітей сприяє як утворенню амілази під шлункової залози, так і її виділенню із слиною. В перші дні і тижні життя амілаза практично відсутня, а слиновиділення незначне, і лише з 3-4 міс. починається секреція амілази і різко зростає слиновиділення.
Відомо, що гідроліз крохмалю відбувається при дії амілаз слини і панкреатичного соку: крохмаль розщеплюється до мальтози і ізомальтози.
Поряд з дисахаридами їжі — лактозою і сахарозою — мальтоза та ізомальтоза на поверхні кишкових ворсинок під впливом дисахаридаз розщеплюються до моносахаридів: глюкози, фруктози і галактози, які підлягають резорбції через клітинну оболонку. Процес резорбції глюкози і галактози зв'язаний з активним транспортом, який полягає у фосфорилюванні моносахаридів і перетворенні їх в глюкозофосфати, а потім в глюкозо-6-фосфат (відповідно галактозофосфати). Така активація відбувається під впливом глюкозо-або галактозокіназ із затратою одного макроергічного зв'язку АТФ. На протилежність глюкозі і галактозі фруктоза резорбується майже пасивно шляхом простої дифузії.
Дисахаридази в кишках плода формуються залежно від терміну гестації, причому раніше підвищується активність мальтази і сахарази (6-8 міс. гестації), пізніше (8-10 міс.) — лактази. Вивчена активність різних дисахаридаз в клітинах слизової оболонки кишки і встановлено, що загальна активність на час народження в результаті дії всіх мальтаз при розщепленні мальтози становить в середньому 246 мкмоль, загальна активність сахарази — 75 мкмоль, ізомальтази — 45 мкмоль, лактази — 30 мкмоль розщепленого дисахарида на 1 г білка за 1 хв. Ці дані викликають великий інтерес у педіатрів, оскільки стає ясно, чому грудна дитина добре перетравлює декстринмальтозні суміші, в той час як лактоза легко викликає діарею.
Відносно низькою активністю лактази в слизовій оболонці тонкої кишки пояснюється той факт, що лактазна недостатність спостерігається частіше, ніж недостатність інших дисахаридаз. Зустрічається як транзиторна мальабсорбція лактози, так і вроджена. Перша її форма обумовлена затримкою дозрівання кишкової лактази, яка з віком дитини зникає. Вроджена ж форма може спостерігатися більш тривалий час, але, як правило, найбільш виражена з народження при грудному вигодовуванні. Це пояснюється тим, що вміст Молочного цукру (лактози) в жіночому молоці майже в 2 рази більший, ніж в коров'ячому. Клінічно у дитини виникає діарея, для якої характерні поряд з частим випорожненням (більше 5 разів на добу) пінисті випорожнення кислої Реакції (рН менше 6). Можуть спостерігатися і симптоми дегідратації, що Проявляються тяжким станом.
Таким чином, вуглеводний обмін у дітей раннього віку характеризується високим засвоєнням вуглеводів (98-99%) незалежно від характеру вигодо- вУвання.
235

Розділ 9
У більш старшому віці відбувається так звана репресія лактази, коли її активність значно знижується. Цим пояснюється те, що більшість людей не переносять молока, в той час як кисломолочні продукти (кефір, ацидофілін, простокваша) засвоюються добре. На лактазну недостатність хворіють близько 75% негрів та індійців, до 90% осіб азіатського походження і 20% кавказців.
Рідше спостерігається вроджена мальабсорбція сахарози і мальтози, яка частіше проявляється у дітей в разі штучного вигодовування, оскільки більшість молочних сумішей збагачується сахарозою, або при включенні до раціону соків, фруктів, овочів, що містять цей дисахарид. Клінічні прояви сахарозної недостатності аналогічні таким при лактозній мальабсорбції.
Описані аналогічні клінічні прояви і при порушенні активування моносахаридів — глюкози і галактози. їх слід відрізняти від випадків, коли в раціоні харчування міститься надто велика кількість цих моносахаридів, які при надходженні до кишок мають високу осмотичну активність, що й викликає надходження до кишок води . Оскільки всмоктування моносахаридів відбувається з тонкої кишки в систему у.роітде, вони в першу чергу надходять до гепатоцитів. Залежно від умов, які визначаються головним чином рівнем глюкози в крові, моносахариди перетворюються в глікоген або залишаються у вигляді моносахаридів і розносяться з кровообігом. У крові дорослих вміст глікогену дещо менший (0,075 — 0,117 г/л), ніж у дітей (0,117 — 0,206 г/л).
Синтез резервного вуглеводу організму — глікогену — відбувається групою різних ферментів, в результаті чого утворюються розгалужені молекули глікогену, що складаються з глюкозних залишків, які сполучуються 1,4 або 1,6 зв'язками (бокові ланцюги глікогену утворюються 1,6 зв'язками). У разі необхідності глікоген знову зможе розщеплюватися до глюкози.
Процес глікогеноутворення у плода і дорослої людини ідентичний, однак існують вікові відмінності в ступені його активності. Так, встановлено, що у плода в останні 2-3 міс. внутрішньоутробного розвитку відбувається дуже активний синтез глікогену. Але вже в перші години після народження майже весь глікоген розщеплюється для забезпечення енерговитрат організму, а його синтез не відбувається фактично до кінця 2-го початку 3-го місяця життя дитини.
Вміст глікогену в печінці в останні тижні вагітності досягає 10% маси органа, але протягом першої доби життя він знижується до 1%.
Таким чином, доношені новонароджені мають досить великі резерви глікогену, які можуть забезпечити майже 20-годинну потребу організму, тоді як недоношені діти мають недостатню кількість глікогену. Причому для дітей раннього віку характерною є недостатня глікогенсинтезуюча зтатність печінки, а також швидке виснаження запасів вуглеводів із печінки.
Крім того, можуть спостерігатися вроджені дефекти ензимних систем, при яких синтез або розпад глікогену в печінці або м'язах може порушуватися. Д° цих станів належить хвороба недостачі резервів глікогену. В її основі лежить
236
Особливості обміну речовин
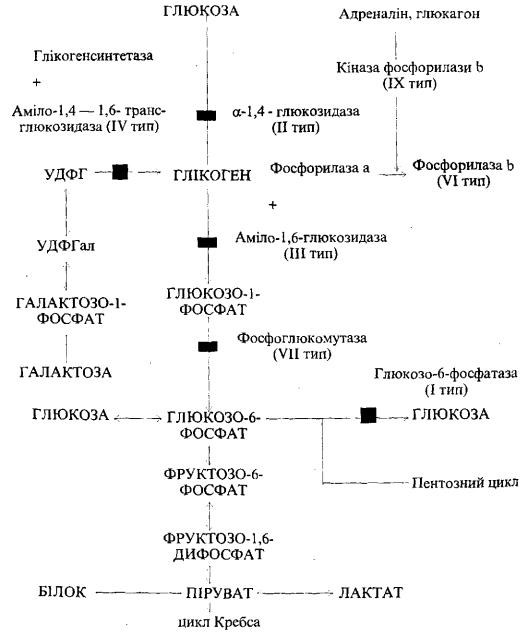
дефіцит ферменту глікогенсинтетази. Рідкість цього захворювання, певно, пояснюється труднощами діагностики і швидким несприятливим наслідком. У новонароджених дуже рано спостерігається гіпоглікемія (навіть в перерві між годуваннями) з судомами і кетозом. Частіше ж в літературі описують випадки глікогенової хвороби, при якій в організмі накопичується глікоген нормальної структури або утворюється глікоген неправильної структури, що нагадує целюлозу (амілопектин). Ця група, як правило, генетично детермі нована. Залежно від дефіциту тих чи інших ферментів, які беруть участь у метаболізмі глікогену, виділяють різні форми, або типи, глікогенозів (рис. 48).
В основі І типу, до якого належить гепаторенальний глікогеноз, або хвороба Гірке, лежить недостатність глюкозо-6-фосфатази. Це найтяжчий варіант серед глікогенозів без структурних порушень глікогену. Захворювання передається за рецесивним типом спадковості. Клінічно проявляється відразу після народження або в грудному віці. Характерна гепатомегалія, яка супро воджується гіпоглікемічними судомами і комою, кетозом; селезінка не збільшена. В подальшому спостерігаються відставання в рості, диспропорція будови тіла (живіт збільшений, тулуб подовжений, ноги короткі, голова велика). В перервах між годуванням відмічаються блідість, пітливість, втрата свідомості як результат гіпоглікемії.
II тип глікогенозу — хвороба Помпе, в основі якої лежить недостатність кислої лізосомальної а-1,4-глюкозидази. Структура глікогену нормальна, але неможливе його р о з щ е п л е н н я . Клінічно проявляєтьс я відразу після народження, і такі діти швидко вмирають . Спостерігаються гепато- і кардіомегамія, м'язова гіпотонія (дитина не може тримати голови, ссати). Розвивається серцева недостатність.
III тип глікогенозу — хвороба Корі — обумовлений вродженим дефектом аміло-1,6-глюкозидази, ферменту, що усуває розгалуження молекули глікогену, і (або) оліго-1,4—1,4-глюканотрансферази. В результаті відкладається патологічний, так званий лімітдекстрин. Передається за аутосомним типом спадковості. Клінічні прояви схожі з такими при І типі (хвороба Гірке), але менш тяжкі. На відміну від хвороби Гірке, це обмежений глікогеноз, який не супроводжується кетозом і тяжкою гіпоглікемією. Глікоген відкладається або в печінці (гепатомегалія), або в печінці і одночасно в м'язах.
IV тип — хвороба Андерсена — обумовлений дефіцитом аміло-1,4 — 1,6- транеглюкозидази, внаслідок чого утворюється глікоген неправильної структури, який нагадує целюлозу (амінопектин). Він є неначе чужорідним тілом. Спостерігаються жовтяниця, гепатомегалія. Формується цироз печінки 3 портальною гіпертензією. Внаслідок цього розвивається варикозне Розширення вен шлунка і стравоходу, розрив яких викликає профузну ^Лункову кровотечу.
V тип — м'язовий глікогеноз, хвороба Мак-Ардла — розвивається у зв'язку 3 Дефіцитом м'язової фосфорилази. Захворювання може проявлятися на 3-му
237
Розділ9_
Рис. 48. Схема обміну глікогену в печінці і порушення активності ферментів при різних типах глікогенозів.
Умовні позначення: УДФГ — уридиндифосфатглюкоза, УДФГал — уридиндифосфатгалактоза
238
Особливості обміну речовин
місяці життя, коли відмічається неспроможність дітей довго ссати грудь, швидка стомлюваність. У зв'язку з поступовим накопиченням глікогену в мускулатурі спостерігається їх псевдогіпертрофія.
VI тип глікогенозу — хвороба Герса — обумовлений дефіцитом печінкової фосфорилази. Структура глікогену нормальна. Відкладання його відбувається в печінці, лейкоцитах та еритроцитах. Клінічно виявляється гепатомегалія, рідше виникає гіпоглікемія і ацидоз. Відмічається відставання в рості. Перебіг більш сприятливий, ніж інших типів. Це найчастіший тип глікогенозу.
Тип VII — недостатня активність фосфофруктокінази. Глікоген нормальної структури, відкладається в м'язах та еритроцитах. Клінічна картина така як при V типі.
Тип VIII виникає в зв'язку з наявністю інгібітора фосфогексоізомерази. Глікоген нормальної структури, відкладається в печінці. Клінічно відмічається відчуття м'язової втоми.
Тип IX — недостатня активність фосфорилазокінази. Глікоген нормальної структури, накопичується в печінці. Клінічні симптоми такі, як при VI типі.
Спостерігаються й інші форми порушення метаболізму, при яких виявляється поліензимний дефект.
Співвідношення інтенсивності процесів глікогенезу і глікогенолізу значною мірою визначає рівень глюкози в крові — глікемію. Ця величина досить постійна. Глікемія регулюється складною системою. Центральною ланкою цієї регуляції є так званий цукровий центр, який потрібно розглядати як функціональне об'єднання нервових центрів, що знаходяться в різних відділах ЦНС, — корі головного мозку, підкірці (сочкувате ядро, смугасте тіло), гіпоталамічній ділянці, подовжньому мозку. П о р я д з цим в регуляції вуглеводного обміну беруть участь багато ендокринних залоз (підшлункова, надниркова, щитовидна).
Одним з показників вуглеводного обміну є вміст глюкози в крові. На момент народження рівень глікемії у дитини становить 70-80% рівня глюкози в крові її матері, що пояснюється вільною трансплацентарною дифузією. У новонарод женого на момент народження рівень глюкози становить 3,33-5,55 ммоль/л (табл. 48).
Проте з перших годин життя (як правило, протягом 3-6 год.) спостерігається значне зниження вмісту глюкози. її рівень знижується до 0,55-1,11 ммоль/л. Причому у деяких дітей рівень глюкози знижується до таких значень, які у Дорослих призводять до виникнення гіпоглікемічної коми. Це пов'язано з двома причинами. Однією з них є досить швидке виснаження запасів глікогену в організмі дитини під час пологів і новонароджений, якого не годують груддю кШька годин після народження, витрачає ці запаси. Швидке виснаження запасів глікогену обумовлене тим, що організм дитини спочатку утилізує вуглеводи, а пізніше жири до тих пір, поки не зможе покрити енергетичні витрати вЖиваннямїжі. Другою причиною гіпоглікемії у новонароджених є недостатня
239
Розділ 9
|
|
|
|
|
Таблиця 48 |
|
Рівень цукру (глюкози) в крові дітей та їх матерів |
|
|
||||
(за методом Хагедорна-Йенсена) |
|
|
|
|
||
|
|
|
|
|
||
Об'єкт |
Середня величина, |
Межі коливань |
|
|||
дослідження |
ммоль/л |
мг% |
|
ммоль/л |
||
Кров матері |
5,16+0,64 |
7 3 - 110 |
|
4,05- 6,10 |
||
Кров з пуповини |
4,75+0,99 |
60100 |
|
3,33 - 5,55 |
||
Кров новонародженого |
|
|
|
|
|
|
через 1 год. |
|
|
|
|
|
|
після народження |
3,10+0,83 |
26 -73 |
|
1,44- |
4,05 |
|
через 24 год. |
3,61+0,77 |
44 -94 |
|
2,44- |
5,22 |
|
через 48 год. |
3,82+0,70 |
42 -84 |
|
2,33- 4,66 |
||
через 72 год. |
4,20+0,89 |
40 - |
100 |
|
2,22- |
5,55 |
Кров дітей 1-12 міс. |
- |
70 -90 |
|
3,89 -5,0 |
||
Кров дітей 2-11 років |
- |
80- |
100 |
|
4,4- 5,55 |
|
Кров дітей 12-14 років |
- |
90- |
120 |
|
5,0- 6,66 |
|
кількість контрінсулярних гормонів. Це підтверджується тим, що адреналін і глюкагон здатні підвищувати рівень глюкози в крові на даний період.
До 5-6-го дня життя вміст глюкози збільшується, але її рівень у дітей залишається відносно нижчим, ніж у дорослих. Підвищення вмісту глюкози в крові дітей після першого року життя хвильоподібне (перша хвиля — до 6 років, друга — до 12 років), що збігається з підсиленим ростом дитини і більш високою концентрацією соматотропного гормону.
Слід пам'ятати, що у дітей до 7-річного віку зберігається схильність до гі поглікемічних реакцій при недостатньому забезпеченні харчування вуглеводами, і тільки після 7 років життя стабілізується регуляція рівня глюкози в крові.
Необхідно підкреслити, що наведені дані рівня цукру в крові отримані за методом Хагедорна-Ієнсена, який є менш точним, порівняно з іншими методами, зокрема глюкозооксидазним або ортотолуїдиновим методом. Якщо рівень глюкози визначається глюкозооксидазним або ортотолуїдиновим методом, то її вміст на 10% нижчий, ніж визначений за методом ХагедорнаІєнсена. Так, у дітей грудного віку рівень глюкози вранці натще за глюкозооксидазним методом становить 2,78-4,4 ммоль/л, у дітей раннього віку
— 3,3-5 ммоль/л і у школярів — 3,3-5,5 ммоль/л.
Рівень глюкози в крові вранці натще у дітей, який перевищує 6,1 ммоль/л (за глюкозооксидазним методом) або 6,66 ммоль/л (за методом ХагедорнаІєнсена) вважають гіперглікемією, а зниження рівня відповідно до 2,5 та 2,8 ммоль/л — гіпоглікемією.
Слід звернути увагу ще на одну особливість вуглеводного обміну, яка може бути причиною низького рівня глюкози. В дитячому організмі послаблені
240