

11 Portopulmonary Hypertension and Hepatopulmonary Syndrome |
|
|
|
|
|
|
|
179 |
|
|||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Chronic Liver Disease Patient with Dyspnea |
|
|
|
|
|
|
|
|
|
|
|
|
||||||
|
|
|
|
|
|
or being evaluated for Liver Transplantation |
|
|
|
|
|
|
|
|
|
|
|
|
||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Echocardiogram |
|
|
Elevated right ventricular |
||||||||||||
|
|
|
Late bubbles in left atrium |
|
|
|
|
|
|
|
|
|||||||||||||
|
|
|
|
|
|
|
|
|
|
|
systolic pressure |
|||||||||||||
|
|
|
|
|
|
|
|
|
No evidence of |
|
|
|
|
|
|
|
|
|
|
|
|
|
||
|
|
Possible HPS |
|
|
|
|
|
PoPH or HPS |
|
|
|
|
|
|
|
|
|
|
|
|
|
|||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Additional Dyspnea Workup (Pulmonary |
|
|
|
Possible PoPH |
|
|
|
|
||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||
|
|
|
Arterial Blood Gason |
|
FunctionTesting,ChestImaging, Laboratory |
|
|
|
|
|
|
|
Right Heart |
|||||||||||
|
|
|
Room Air |
|
|
|
Testing, etc.) ifneeded |
|
|
|
|
|
|
|
|
|||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Catheterization |
||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||
|
|
|
|
|
|
|
Cleared for liver transplantation from a |
mPAP>20mmHg |
|
|
|
|
|
|
||||||||||
|
|
|
|
|
|
|
pulmonary vascular disease standpoint |
PCWP ≤ 15mmHg |
|
|
|
|
|
|
||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
PVR ≥ 3WU |
|
|
|
|
|
|
|||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Confirmed PAH |
|
|
mPAP ≤ 20mmHg |
||||||
|
|
|
|
|
|
|
|
|
PaO2 and A-agradient |
|
Workup for other causes of PAH |
|
or |
|||||||||||
|
|
|
|
|
|
|
|
|
|
|
mPAP > 20mmHg |
|||||||||||||
|
|
|
|
|
|
|
|
|
|
(V/Q scan, laboratory testing, etc.) |
|
|||||||||||||
|
|
|
PaO2 < 80mmHg* |
|
|
within normal limits |
|
|
PVR < 3WU |
|||||||||||||||
|
|
|
A-agradient ≥ 15mmHg* |
|
|
|
|
|
Consideration for targeted |
|
|
|
|
|
|
|||||||||
|
|
|
|
|
|
|
|
|
|
|
|
pulmonary vasodilator therapy |
|
|
|
|
|
|
||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||
|
|
|
Confirmed HPS |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Does not meet diagnostic |
|
|
|
|
|
|
|
|
|
Not PoPH |
|
|
|
||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||
|
Expedited Liver Transplantation Workup |
|
|
criteria for HPS |
|
|
|
|
|
Evaluation for PH (if present) by |
|
|
||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||||
|
Supplemental Oxygen Therapy if needed |
|
|
Consider serial blood gas test monitoring |
|
pulmonary vascular disease specialist |
|
|
||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
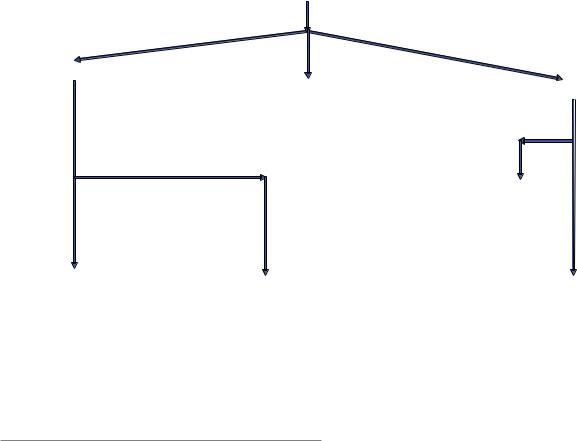
Fig. 11.1 Algorithm for the evaluation of the dyspneic chronic liver disease patient. PH pulmonary hypertension, HPS hepatopulmonary syndrome, PoPH portopulmonary hypertension, A-a gradient alveolararterial oxygen gradient, PaO2 partial pressure of oxygen, V/Q
ventilation-perfusion testing, mPAP mean pulmonary arterial pressure, PCWP pulmonary capillary wedge pressure, PVR pulmonary vascular resistance, WU woods units
Portopulmonary Hypertension (PoPH)
Epidemiology and Risk Factors
Portopulmonary hypertension is a pulmonary vascular complication of liver disease, defned by the presence of pulmonary arterial hypertension (PAH) in the setting of portal hypertension [5–7]. Although virtually all patients with liver cirrhosis also suffer from portal hypertension, it is important to note that portal hypertension can also rarely occur in the absence of liver cirrhosis, and that PoPH can occur in patients with non-cirrhotic portal hypertension [8]. Portal hypertension is identifed either directly by catheter-based portal vein hemodynamic measurements, or inferred by the presence of splenomegaly, thrombocytopenia, portosystemic shunts, esophageal varices, or portal vein abnormalities [5–7]. PAH is best defned by right heart catheterization (RHC) hemodynamic measurement as a mean pulmonary arterial pressure of >20 mmHg at rest, with a pulmonary capillary wedge pressure ≤15 mmHg, and a pulmonary vascular resistance of at least 3 Woods Units (240 dynes/s/cm−5) [1, 2]. Identifying PAH in a patient with portal hypertension, and excluding
other causes of PAH (such as connective tissue disease or chronic thromboembolic disease) confrms the diagnosis of PoPH.
Estimates for the prevalence of PoPH vary widely, in part due to challenges in diagnosis and classifcation [5–7, 9–11]. Among all patients with portal hypertension and chronic liver disease, it is estimated that between 1% and 6% of patients carry a diagnosis of PoPH. In patients who undergo comprehensive diagnostic testing for liver transplant evaluation, the prevalence of PoPH ranges from 5% to 8.5% of patients. PoPH accounts for between 5% and 10% of all PAH diagnoses. From a large study of patients in the United Kingdom, the prevalence of PoPH was estimated at 0.85 per million persons [9]. The diagnosis is typically made in the fourth to ffth decade of life, and often 4–7 years after portal hypertension is diagnosed. Female sex and autoimmune hepatitis are known to be associated with the presence of PoPH, and hepatitis C patients are less likely to develop PoPH [9–13]. There is no known association between either the degree of portal hypertension or liver cirrhotic dysfunction and either the presence and severity of PoPH.
Данная книга находится в списке для перевода на русский язык сайта https://meduniver.com/

180 |
A. Jose et al. |
|
|
Molecular Pathogenesis
The molecular pathogenesis in PoPH is incompletely understood, but vasoactive mediators that affect the pulmonary vasculature are believed to play a central role (Fig. 11.2). Patients with PoPH demonstrate the same histopathological changes of pulmonary artery intimal fbrosis, smooth muscle medial hypertrophy, in-situ thrombosis, and plexiform lesions that are present in other patients with PAH (Fig. 11.3) [5–7]. Endothelin-1 (ET-1), a vasoactive peptide secreted by vascular endothelial and activated stellate cells, likely plays a central role in PoPH disease pathogenesis. ET-1 is a potent systemic and pulmonary vasoconstrictor that acts on hepatic stellate and endothelial cells to regulate blood ow, extracellular matrix turnover, and fbrosis in chronic liver disease. ET-1 is pleiotropic, and its binding to the ET-A receptor promotes vasoconstriction and smooth muscle cell prolifera-
a
tion, and to the ET-B receptor contributes to pulmonary vascular remodeling and endothelial-to-mesenchymal cell transformation in the pulmonary vasculature [5, 14, 15]. Signifcantly higher pulmonary arterial concentrations of ET-1 have been observed in PoPH as compared patients with liver cirrhosis without PoPH [5, 11, 16, 17]. Multiple other vasoactive factors, believed to promote disease pathogenesis in other forms of PAH, have also been implicated in PoPH pathogenesis. These candidate mediators, including interleu- kin-6, thromboxane B-1, serotonin, prostaglandins, and nitric oxide, are believed to interact with ET-1 to promote in ammation, angiogenesis, and pulmonary vascular remodeling [5, 11]. Macrophages are likely also key effectors in disease pathogenesis, and levels of macrophage migration inhibitory factor have been found to be signifcantly higher in cirrhotic patients with PoPH as compared to those without PoPH [18].
b
c
Fig. 11.2 Working Hypothesis for the Molecular Pathogenesis of PoPH. Endothelin-1 (ET-1) derived from hepatic endothelial and activated stellate cells induces these cells to alter regulation of hepatic blood ow, in ammation, and extracellular matrix (ECM) turnover (a). ET-1 is also released into systemic circulation, where it acts in concert with circulating thromboxane B-1, nitric oxide, interleukin-6 (IL-6),
serotonin, prostaglandins, and in the presence of low BMP-9 levels (due to endoglin binding and sequestration), to activate pulmonary macrophages and promote endothelial-to-mesenchymal cell transition (EMT) (b), smooth muscle cell (SMC) proliferation (c), and pulmonary vascular remodeling characteristic of PoPH

11 Portopulmonary Hypertension and Hepatopulmonary Syndrome |
181 |
|
|
Fig. 11.3 Representative histopathology of lung explant from a 37-year-old woman with primary biliary cirrhosis who underwent heart–lung–liver transplant, showing pulmonary artery medial hypertrophy, intimal fbrosis, and plexiform lesions (Hematoxylin and Eosin stain, black arrow)
The bone morphogenic protein system has also recently been implicated in the pathogenesis of PoPH [19–22]. Bone morphogenic protein 9 (BMP-9), encoded by the growth differentiation factor 2 gene (GDF2), is a protein member of the transforming growth factor beta superfamily produced in the liver. BMP-9 levels were found to be signifcantly decreased in patients with PoPH as compared to patients with other forms of PAH, or those with non-PoPH liver cirrhosis. This difference persists even after adjustment for age, gender, and severity of liver disease as measured by the Model for End-Stage Liver Disease (MELD) score. Additionally, in a mouse carbon tetrachloride model of liver cirrhosis, while binding of BMP-9 to hepatic stellate cells induces hepatocyte proliferation and mesenchymal transition of hepatic stellate cells, blockade of BMP-9 instead promotes development of pulmonary vascular disease following exposure to hypoxemic conditions [19]. A large study of gene polymorphisms in PoPH and non-PoPH cirrhotic liver disease patients did not identify informative variants in GDF2, bone morphogenic protein type 2, or endoglin (the circulating ligand trap for BMP-9) in PoPH patients, however, highlighting the substantial remaining gaps in our understating of PoPH disease pathogenesis [23]. Recently, single-cell RNA-sequencing techniques have shed new light on the cellular transcriptomics that promote progression from fbrosis to cirrhosis in the liver and show promise in identifying the molecular mechanisms that underlie PoPH disease pathogenesis as well [24].
Screening andDiagnosis
Screening for PoPH is currently recommended for all patients undergoing LT evaluation (Fig. 11.1) [1, 3, 5–7, 11, 25, 26]. Clinical manifestations of PoPH are nonspecifc and often diffcult to distinguish from underlying portal hypertension and chronic liver disease. The most common symptom is dyspnea, but fatigue and weakness are also common. Physical examination will typically demonstrate the characteristic fndings of portal hypertension including peripheral edema, abdominal ascites, a pulsatile liver, and the stigmata of cirrhosis (spider angiomas, scleral icterus, jaundice, and palmar erythema) (Fig. 11.4). In the advanced stages of PoPH, patients can present with signs and symptoms of right heart failure, including syncope, orthopnea, and a physical examination demonstrating an elevated jugular venous pulse, a prominent and split second heart sound, a tricuspid regurgitant murmur, and a palpable right ventricular heave. Electrocardiogram testing can show rightaxis deviation, electrical evidence of right ventricular and right atrial enlargement, and occasionally a right bundle branch block pattern. Thoracic imaging (chest radiography and computed tomography) is nonspecifc, occasionally demonstrating the presence of an enlarged right ventricle and a dilated central pulmonary artery suggestive of underlying pulmonary vascular disease. Pulmonary function testing will occasionally show a decreased diffusion capacity for carbon monoxide, but this is not diagnostic of PoPH. Serum levels of brain natriuretic peptide, the most widely used biomarker in PAH, are known to be elevated in portal hypertension and chronic liver disease, and are of limited value in screening patients with these conditions for underlying PoPH [27].
Resting transthoracic echocardiographic (TTE) imaging is the optimal screening tool for PoPH and is recommended in all patients undergoing evaluation for LT [3]. Elevations in the estimated right ventricular systolic pressure (RVSP) can identify patients at risk for PoPH who warrant further workup with diagnostic RHC testing. PoPH can rapidly develop in patients with underlying portal hypertension, and therefore, serial TTE screening is recommended for all liver transplant candidates, to avoid the morbidity and mortality associated with performing LT in a patient with undiagnosed PoPH. Although RVSP cutoffs vary across institutions, it is generally accepted that an RVSP lower limit on TTE of 30 mmHg is suffcient (>95% negative predictive value) to exclude clinically signifcant PoPH. This test value is tempered by a limited specifcity (59–77%) for echocardiography in accurately identifying PoPH, as the correlation
Данная книга находится в списке для перевода на русский язык сайта https://meduniver.com/

182 |
A. Jose et al. |
|
|
Fig. 11.4 Characteristic physical exam fndings of chronic liver cirrhosis including palmar erythema and jaundice (left panel) and spider angiomas (right panel)
between RVSP on TTE and pulmonary arterial pressures on RHC is moderate at best (linear correlation strength ~0.46– 0.78) [3, 28–30].
As with all forms of PAH, the diagnosis of PoPH requires invasive RHC hemodynamic measurements [1, 2, 5–7, 31]. Due to abnormal vascular morphology and alterations in compensatory mechanisms regulated by the estrogen axis and the renin–angiotensin–aldosterone system, the majority of patients with portal hypertension and liver cirrhosis will demonstrate a hyperdynamic circulation, with elevated pulmonary arterial pressures (and elevated RVSP measurements on TTE) as a consequence of an increase in cardiac output, but with normal pulmonary vascular resistance [32, 33]. Patients with PoPH, by contrast, will demonstrate elevations in pulmonary arterial pressure that are in excess of what can be accounted for on the basis of increased cardiac function, resulting in a markedly abnormal elevation in calculated pulmonary vascular resistance. The diagnosis of PAH requires a mean pulmonary arterial pressure >20 mmHg at rest, with a pulmonary capillary wedge pressure ≤15 mmHg, and a pulmonary vascular resistance of at least 3 Wood Units (240 dynes/s/cm−5) [1, 2]. Of note, many chronic liver disease patients will have a pulmonary vascular resistance that does not meet this threshold but still display abnormally elevated pulmonary arterial pressures. It is unclear what the signifcance of abnormally elevated pulmonary arterial pressures is in this patient population, but increasing evidence suggests that these patients are prone to worse outcomes while awaiting transplantation and have increased mortality post-LT [5–7, 13, 31, 34].
PoPH Treatment
The treatment for PoPH can be divided into two categories: liver transplantation and medical therapy with targeted pulmonary vasodilators. Unfortunately, survival and outcomes in PoPH remain poor regardless of treatment [4–7, 9, 11, 13, 35–38]. A large retrospective study of PoPH patients from the United Kingdom estimated 1-year survival at 85%, and 5-year survival at 35%, even with medical therapy [9]. These estimates aligned with other reports of 1-year survivals between 86% and 91% but are signifcantly worse than the more commonly cited 67–70% 5-year survival in treated PoPH. Survival in PoPH, similar to other forms of PAH, is highly correlated with the degree of right ventricular failure, as measured by RHC cardiac output, cardiac index, and pulmonary vascular resistance. In PoPH, survival is also associated with the degree of liver disease, as measured by the MELD score. Although PoPH is not an indication for LT, given the high morbidity and mortality in this population, MELD exception points are granted for a diagnosis of PoPH, and LT is generally offered for PoPH patients who are able to achieve a reduction in mean pulmonary arterial pressure to ≤35 mmHg on RHC with a pulmonary vascular resistance of <3 Wood Units [3, 34]. As PoPH can progress rapidly, MELD exception points must be updated every 6 months with repeat RHC testing. Therapeutic reduction in pulmonary vascular pressures improves outcomes of LT. A retrospective single center study of PoPH patients undergoing liver transplantation indicated 100% mortality in those with mean pulmonary arterial pressure of >50 mmHg, and no mortality if mean pul-
11 Portopulmonary Hypertension and Hepatopulmonary Syndrome |
183 |
|
|
monary arterial pressure was less than 35 mmHg, highlighting the importance of pulmonary hemodynamics in facilitating safe LT in these patients [39]. As a consequence of this study, PoPH with a mean pulmonary arterial pressure >45 mmHg is considered an absolute contraindication to LT at most centers.
Even when the above hemodynamic criteria are met, LT in PoPH is still considered high risk and is associated with a signifcant morbidity and mortality post-transplant. A review of the Multicenter Liver Transplant Database indicated that the post-transplant hospital mortality in PoPH patients undergoing LT was 36%, although these data preceded the modern era of targeted PAH treatment for PoPH, and more recent estimates [37] for post-LT survival in PoPH range from 77% to 91% at 1 year, and 67–80% at 5 years. It is generally accepted that the majority of PoPH patients who undergo LT will experience an improvement in the severity of their pulmonary vascular disease, and some are cured of PoPH post-transplant, but the factors that predict a favorable response to LT are still unknown [40–47]. Additionally, relapse of PoPH has been reported after an initially favorable outcome from LT, and therefore, serial TTE screening is recommended following transplantation [3–5, 11].
Medical therapy represents the other pillar of treatment for PoPH, which complements and facilitates LT. The cornerstone of medical therapy in PoPH mirrors that of other types of PAH and is centered on the use of targeted pulmonary vasodilator therapy to reduce pulmonary vascular resistance [48–60]. As PoPH patients are generally excluded from most PAH clinical trials, the limited effcacy data available in PoPH is primarily derived from case series and small cohort studies, with few randomized controlled studies to support use of these agents. The earliest studied agent for PoPH was epoprostenol, a synthetic analog of a naturally occurring prostacyclin, which has potent vasodilatory properties as well as anti-in ammatory and anti-platelet effects. Data for the use of epoprostenol comes from case series and retrospective cohort data, in which administration of epoprostenol was shown to result in signifcant improvements in pulmonary vascular hemodynamics (mean pulmonary arterial pressure, cardiac output, cardiac index, and pulmonary vascular resistance), and facilitated successful LT in some patients [54, 58–60]. Given the signifcant benefts of epoprostenol in other forms of PAH, and the fact that epoprostenol remains the only therapeutic agent to demonstrate a mortality beneft in PAH, it is accepted that intravenous epoprostenol is the preferred therapeutic for PoPH [61, 62].
Oral pulmonary vasodilator medications have also demonstrated clinical beneft in PoPH and remain an important
part of the medical armamentarium to treat this condition. Oral phosphodiesterase 5 inhibitor medications, sildenafl and tadalafl, have demonstrated beneft in case series and cohort studies of PoPH [52–57]. Sildenafl monotherapy results in improvements in walk distance, functional capacity, circulating natriuretic peptide levels, pulmonary vascular hemodynamics (mean pulmonary arterial pressure, pulmonary vascular resistance, and cardiac output), and has facilitated successful LT in PoPH patients. A newer class of medications, soluble guanylate cyclase stimulators (riociguat), has also shown effcacy in improving pulmonary vascular hemodynamics and functional capacity in PoPH [51]. Given the potential role of ET-1 in driving PoPH disease pathogenesis, endothelin receptor antagonist medications (ambrisentan, macitentan) have been carefully studied for effcacy in PoPH. Notably, endothelin receptor antagonists are the only medications to show beneft in PoPH in a randomized controlled trial setting and have been successfully used either alone or in combination with phosphodiesterase 5 inhibitors in patients with PoPH [48–50, 55]. The recently published PORTICO study, the largest clinical trial to date in PoPH patients, randomized 85 PoPH patients to either macitentan or placebo, and demonstrated a signifcant improvement in pulmonary vascular resistance, mean pulmonary artery pressure, and cardiac index on RHC after macitentan treatment for 12 weeks [48]. Smaller case series and open-label studies have shown similar improvements in pulmonary vascular hemodynamics following endothelin receptor antagonist therapy in PoPH, and despite the hepatic metabolism of this class of medications, only a minority of patients experienced liver function abnormalities signifcant enough to warrant medication cessation. Notably, all targeted pulmonary vasodilator medications are hepatically metabolized except epoprostenol (which is rapidly hydrolyzed in plasma), and therapeutic plans should anticipate the potential for higher drug concentrations and longer half-lives in PoPH compared to other PAH subtypes [63]. Other considerations when using these medications include the increased peripheral edema with endothelin receptor antagonists (which can be dose limiting in patients with pre-existing ascites and edema from portal hypertension), and the enhanced action of phosphodiesterase 5 inhibitors and prostacyclin analogs when co-administered with ethanol (which can occasionally be seen in patients with underlying alcoholic cirrhosis).
In addition to targeted vasodilator therapy for PoPH, there are other therapeutic interventions that merit discussion. Patients with portal hypertension are often treated with beta blocker medications to decrease portal pressure and prevent
Данная книга находится в списке для перевода на русский язык сайта https://meduniver.com/