

82 |
H. J. Lachmann and J. H. Pinney |
|
|
Systemic Amyloidosis Complicating
Respiratory Diseases
Systemic AA Amyloidosis
Systemic AA amyloidosis is a potential complication of any disorder associated with a sustained acute-phase response, and the list of chronic infammatory, infective or neoplastic disorders that can underlie it is almost without limit (Table 6.2). Biopsy and postmortem series suggest that the prevalence of AA amyloid deposition in patients with chronic infammatory diseases is between 3.6% and 5.8%, though a smaller proportion of patients have clinically signi cant amyloidosis [38–40]. The amyloid brils are derived from cleavage fragments of the circulating acute-phase reactant, SAA [41]. SAA is an apolipoprotein of high-density lipoprotein (HDL), which is synthesised by hepatocytes under the transcriptional regulation of cytokines including interleukin (IL)-1, IL-6 and tumour necrosis factor-α (TNF-α) [42]. In health, the circulating concentration of SAA is around 1 mg/L, but this can rise by more than a 1000-fold in the presence of infammation. The circulating concentration of SAA tends to be parallel to that of the much more frequently measured C-reactive protein (CRP). A sustained high plasma level of SAA is a prerequisite for the development of AA amyloidosis, but the reason why amyloidosis develops in only a small proportion of cases remains unclear. AA amyloidosis can present anytime between childhood and old age with a median age at presentation of 48 years in the UK. It is slightly more common in men and, although the disease can develop extremely rapidly, the median latency between presentation with a chronic infammatory disorder and clinically signi cant amyloidosis is almost two decades [43].
Table 6.2 Conditions with respiratory manifestations associated with systemic AA amyloidosis
Chronic infections |
Neoplasia |
Bronchiectasis |
Adenocarcinoma of the lung |
Q fever |
Carcinoid tumour |
Subacute bacterial endocarditis |
Castleman’s disease |
|
|
Tuberculosis |
Hodgkin disease |
|
|
Immunode ciency states |
Mesothelioma |
|
|
Common variable |
Infammatory arthritis |
immunode ciency |
|
Cyclic neutropenia |
Adult Still’s disease |
Hyperimmunoglobulin M syndrome |
Ankylosing spondylitis |
|
|
Hypogammaglobulinaemia |
Rheumatoid arthritis |
|
|
Sex-linked agammaglobulinaemia |
Systemic vasculitis |
|
|
HIV/AIDS |
Behcet’s disease |
Other conditions predisposing to |
Systemic lupus |
chronic infections |
erythematosus |
Cystic brosis |
Others |
Kartagener syndrome |
SAPHO syndrome |
|
|
Quadriplegia |
Sarcoidosis |
|
|
Sickle cell anaemia |
Sinus histiocytosis with |
|
massive lymphadenopathy |
The most common respiratory disease underlying AA amyloidosis in the United Kingdom is bronchiectasis, accounting for 5% cases. A study of 16 patients with end-stage renal failure secondary to AA amyloidosis due to bronchiectasis in Turkey reported the mean duration of bronchiectasis to be 22.18 years, with a wide range of ±12.02 years. The mean age at presentation was 50.6 ± 13.5 years. Eight cases (50%) had cystic bronchiectasis and four of these eight patients died from suppurative pulmonary infections. The other eight patients had chronic brotic changes, and four of these were considered to be the sequelae of previous tuberculosis (TB) infections [44]. The prevalence of bronchiectasis is falling due to earlier treatment of necrotising pneumonia and prevention of pulmonary infections via routine immunisation programmes; hopefully, this will also lead to a reduction of patients with AA amyloidosis from this cause.
Lung neoplasia including Castleman’s tumours, lymphomas and adenocarcinomas account for 3% of cases of AA amyloidosis. Castleman’s disease [45], or angiofollicular lymph node hyperplasia, is a rare B-cell lymphoproliferative disorder characterised by giant hyperplastic lymph node follicles, capillary proliferation and plasma cell in ltration, and it is often associated with marked constitutional symptoms. It comprises solitary and multicentric forms, and there are hyaline vascular and plasma cell variants histologically [46]. Multicentric disease, commonly of the plasma cell type, usually has an aggressive and rapidly fatal course. Unicentric disease tends to occur in younger patients and is of the hyaline vascular type in more than 70% of cases and of the plasma cell type or mixed histology in the remainder [47, 48]. Most solitary tumours occur within the mediastinum and consist of a dominant mass surrounded by multiple enlarged lymph nodes, which, histologically, may appear merely reactive. Constitutional symptoms including night sweats, fever and weight loss are common and laboratory abnormalities including anaemia, elevation of the erythrocyte sedimentation rate (ESR) and polyclonal hypergammaglobulinaemia are almost universal. Acquired systemic amyloidosis is a recognised rare complication of all forms of angiofollicular lymph node hyperplasia and is usually of the systemic AA type, occurring as a result of the persistent acute-phase response [49–52]. In Castleman’s disease, there is production of IL-6 by the tumour and anti-IL-6 therapies can be highly effective [51–56]. In other lymphomas, the exact pathophysiology resulting in a systemic acute-phase response is less well-understood [57]. A surgical resection of unicentric tumours can result in complete remission and excellent long-term outcome [49].
Other purely respiratory causes of AA amyloidosis are now fairly rare in the UK, although in the rst half of the last century, tuberculosis was common, and this remains the case in parts of the developing world and is likely to be underreported [58, 59]. Other rare associations include cystic brosis [60, 61], sarcoidosis [62] and Kartagener syndrome [63].
6 Amyloidosis and the Lungs and Airways |
83 |
|
|
AA amyloidosis usually presents with proteinuria, which can be heavy. Progressive renal dysfunction follows, often accompanied by nephrotic syndrome [64]. Splenic amyloid deposits are almost universally present but are often asymptomatic and frequently impalpable. Hepatic involvement and autonomic neuropathy are well-recognised in advanced disease. Cardiac amyloidosis is extremely rare, occurring in less than 2% of cases. Respiratory tract involvement has not been a clinical feature among more than 400 patients with systemic AA amyloidosis evaluated in our unit, and although there have been a few reports of systemic AA amyloidosis affecting the lungs, bril typing was generally imperfect [65], and all studies in which the bril protein has actually been sequenced have been identi ed to be of the AL type.
A diagnosis of amyloidosis relies on a high index of clinical suspicion. The most effective form of basic screening in medical or respiratory practice is to target patients at a risk of developing AA amyloidosis with ongoing poorly controlled infammation and to perform urinalysis on each clinic attendance. More than 95% of patients with AA amyloidosis will have signi cant proteinuria on dipstick testing, which should prompt investigation. The prognosis of AA amyloidosis depends on the degree of renal dysfunction at presentation and whether the underlying chronic infammatory disease can be effectively suppressed so that the median plasma SAA is maintained below 10 mg/L. When the supply of thebril precursor protein is substantially reduced for sustained periods, AA amyloid deposits frequently regress and renal function can improve [43, 64, 66]. If the acute-phase response continues unabated, then progressive amyloid deposition often results in end-stage renal failure. In individuals who present with advanced renal disease, even complete suppression of their infammatory disease may not be suf cient to preserve their renal function and in all cases renal deterioration is accelerated by hypertension. Treatment depends on the underlying diagnosis and may include surgery for cytokine secreting tumours or localised bronchiectasis, long- term antimicrobials and postural drainage for chronic infections associated with structural lung problems in cysticbrosis or Kartagener and immunosuppression in infammatory diseases such as sarcoidosis.
Almost 40% of patients with AA amyloidosis eventually develop dialysis-dependent renal failure. Renal outcomes on dialysis are equivalent to those of age-matched non-diabetic patients on the end-stage programme with a median survival of 53 months. Mortality is higher in the rst year, and this has been attributed to ongoing heavy urinary protein losses and increased risk of sepsis [67, 68]. Nephrotic syndrome is a major risk factor for sepsis, particularly in patients predisposed to infection, such as those with bronchiectasis, in whom the risk of further infection is even greater and outcome may well be poor. A minority of patients go on to receive renal transplants [68–70]. The published outcomes are rather variable, but our series of almost 40 highly selected
patients with well-suppressed SAA levels had a 5-year graft survival of 82%.
Systemic AL Amyloidosis
This is the most common type of systemic amyloidosis accounting for more than 60% of cases [71] and may potentially occur in association with any form of monoclonal B-cell dyscrasia. The precursor proteins are monoclonal immunoglobulin light chains and generally consist of the whole or part of the variable (VL) domain [72].
A number of conditions localised to the thoracic cavity can underlie systemic AL amyloidosis. An isolated plasmacytoma presenting as a chest mass can secrete enough monoclonal-free immunoglobulin light chains into the circulation to produce systemic AL amyloid deposits [73] (Fig. 6.4a, b). Castleman’s tumours, both unicentric and multicentric, can be associated with monoclonal immunoglobulin light-chain production and are a rare cause of AL amyloidosis [74]. The most common condition managed by respiratory physicians, which can cause both systemic and respiratory localised AL amyloid deposits, is Sjögren’s syndrome, which is discussed later.
A degree of amyloid deposition is seen in up to 15% of patients with myeloma, but the vast majority, more than 80%, who present with clinically signi cant AL amyloidosis have an extremely low grade and otherwise ‘benign’ monoclonal gammopathies [75]. AL amyloidosis usually presents over the age of 50 years, although it can occur in young adults [75]. Clinical manifestations are extremely variable since almost any organ other than the brain can be directly involved [76]. Although speci c clinical features can be strongly suggestive of AL amyloidosis (Table 6.3), and multiple vital organ dysfunction is common, many patients present with non-speci c symptoms such as malaise and weight loss. The outlook of untreated AL amyloid is far worse than that of the AA type, with a 5-year survival of approximately 10% and a 10-year survival of less than 5% [75]. Most affected individuals eventually die of heart failure, uraemia or autonomic failure.
In most cases, there is substantial histological cardiac involvement, and restrictive cardiomyopathy is the presenting feature in up to one-third of patients and ultimately the cause of death in one-half [77]. Renal involvement is frequent in AL amyloidosis and presents in the same manner as renal AA amyloidosis [78]. Gut involvement can cause motility disturbances (often secondary to autonomic neuropathy), malabsorption, perforation, haemorrhage or obstruction [79]. Peripheral neuropathy occurs in onefth of cases and typically presents with a painful sensory polyneuropathy, followed later by motor de cits [75]. Autonomic neuropathy causing orthostatic hypotension, impotence and gastrointestinal disturbances may occur in isolation or with a peripheral neuropathy [76].
Данная книга находится в списке для перевода на русский язык сайта https://meduniver.com/
84 |
H. J. Lachmann and J. H. Pinney |
|
|
a |
b |
c
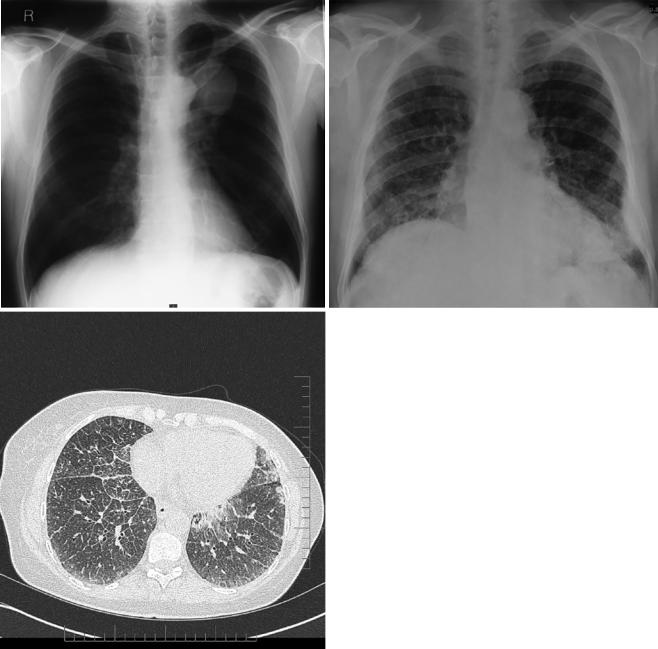
Fig. 6.4 (a) A CXR demonstrating a mass in the left upper lobe; this was diagnosed as a plasmacytoma with associated AL amyloid deposition following a biopsy. (b) A posterior whole-body scintigraphic image from the same patient obtained following an intravenous injection of
123I-human SAP, showing abnormal uptake into the amyloid deposits within the plasmacytoma and deposition in the spleen. High resolution CT of the chest on the same patient con rming intersitial in ltration (c)
Although microscopic deposits of amyloids are universally present in the lungs, in the vast majority of cases, dyspnoea is secondary to cardiac involvement. The chest radiograph is usually normal in systemic AL amyloidosis but may demonstrate a diffuse alveolar septal pattern, and this can be associated with pulmonary hypertension and respiratory failure [80]. When pulmonary symptoms do develop, the outlook is grave and responds poorly to therapy [80, 81]. Patients are often highly dyspnoeic, which may be further compounded by amyloid-induced cardiac dysfunction. The
pathophysiology is due to deposition of amyloid brils within the small airways and the capillary alveolar membrane, leading to impaired gas exchange and respiratory failure detectable by decreased carbon monoxide diffusion capacity. With more widespread involvement, a restrictive defect can occur similar to that seen in pulmonary brosis. Pulmonary function testing provides a quantitative means of both assessing a patient’s baseline dysfunction and tracking the progression or response to treatment. Lung function tests may show a restrictive pattern and more rarely reduced gas transfer [82].