


Amyloidosis and the Lungs and Airways |
6 |
|
|
Helen J. Lachmann and Jennifer H. Pinney |
|
Introduction
Amyloids occur due to the deposition of soluble plasma proteins within the extracellular space in an abnormal insolublebrillar form. Amyloidosis describes a group of diseases caused by the resulting disruption of the tissue structure and organ function. The diagnosis is often made late in the disease course, frequently as an unexpected histological nding when a failing organ is biopsied. It may be either acquired or inherited, and at least 30 different proteins can form amyloidbrils in humans [1] (Table 6.1). In the right conditions, almost any polypeptide chain can be driven towards misfolding and aggregation but relatively few proteins are amyloidogenic in vivo. There are essentially three circumstances in which amyloid deposition occurs. The rst is when there is a sustained, abnormally high abundance of proteins that are normally present at low levels, such as serum amyloid A protein (SAA) in chronic infammation or beta-2 microglobulin in chronic renal failure. The second is when there is normal abundance of a normal but inherently amyloidogenic protein over an extremely prolonged period, such as transthyretin. The third situation is the presence of an abnormal protein, which is markedly amyloidogenic, such as monoclonal immunoglobulin light chains in AL amyloidosis and genetic variants of transthyretin, apolipoprotein AI, brinogen Aα chain, etc. in autosomal dominant hereditary amyloidosis.
The ultrastructural morphological and histochemical properties of all amyloid brils, regardless of the precursor
H. J. Lachmann (*)
UK National Amyloidosis Centre, Division of Medicine, University College London and Royal Free London NHS Foundation Trust, London, UK
e-mail: h.lachmann@ucl.ac.uk; helen.lachmann@nhs.net
J. H. Pinney
Department of Renal Medicine, University Hospital Birmingham NHS Foundation Trust, Birmingham, UK
protein type, are remarkably similar. Diffraction studies of amyloid brils have con rmed that they all share a common core structure consisting of anti-parallel β-strands lying perpendicular to the long axis of the brils. This extremely abnormal, highly ordered conformation underlies the distinctive physicochemical properties of amyloidbrils. The brils are relatively stable and are resistant to proteolysis. All amyloid brils possess the ability to bind molecules of the dye Congo Red in a spatially organised manner, which results in a pathognomonic apple-green birefringence when viewed under crossed polarised light. Amyloid deposits also always contain the normal plasma glycoprotein, serum amyloid P component (SAP) as a non-brillar constituent. The universal presence of SAP in amyloid deposits refects its speci c binding to an as yet uncharacterised ligand common to all amyloid brils, which forms the basis for diagnostic scintigraphic imaging of amyloids with radiolabelled SAP [2].
The phenotypes associated with amyloid deposition are diverse, ranging from an asymptomatic, small, localised deposit to a systemic, rapidly lethal multi-system disease [3]. Clinically important amyloid deposits accumulate in the extracellular space, progressively disrupting the structure, integrity and function of the tissues and organs. The natural history of amyloidosis is usually of progressive accumulation, although as amyloid deposits are constantly turned over, clinical progression refects the fact that the brillar deposits are laid down faster than they are cleared away [4]. Amyloid deposits can therefore regress if this balance is tipped in favour of clearance; current treatment strategies have focused on reducing the supply of brils by halting the production of the culpable plasma protein. Although treatment is not always effective as many of the conditions that underlie systemic amyloidosis are progressive and unremitting, there are numerous reports describing regression of amyloidosis when associated infammatory and other diseases have been successfully treated.
© Springer Nature Switzerland AG 2023 |
77 |
V. Cottin et al. (eds.), Orphan Lung Diseases, https://doi.org/10.1007/978-3-031-12950-6_6 |
|
Данная книга находится в списке для перевода на русский язык сайта https://meduniver.com/

78 |
|
H. J. Lachmann and J. H. Pinney |
|
||
Table 6.1 Classi cation of the more common types of systemic amyloidoses in humans |
||
|
|
|
Type |
Fibril protein precursor |
Clinical syndrome |
AA |
Serum amyloid A protein |
Reactive systemic amyloidosis associated with chronic |
|
|
infammatory diseases |
AL |
Monoclonal immunoglobulin light chains |
Systemic amyloidosis associated with monoclonal |
|
|
plasma cell dyscrasias |
|
|
|
AH |
Monoclonal immunoglobulin heavy chains |
Systemic amyloidosis associated with monoclonal |
|
|
plasma cell dyscrasias |
Aβ2M |
Normal plasma β2-microglobulin |
Periarticular and, occasionally, systemic amyloidosis |
|
|
associated with long-term dialysis |
Aβ2M |
Variant β2-microglobulin |
Autosomal dominant hereditary systemic amyloidosis |
ATTR |
Normal plasma transthyretin |
Wild-type systemic TTR amyloidosis with prominent |
|
|
cardiac involvement |
|
|
|
ATTR |
Genetically variant transthyretin |
Autosomal dominant systemic amyloidosis |
|
|
Familial amyloid polyneuropathy or cardiomyopathy |
ACys |
Genetically variant cystatin C |
Autosomal dominant Systemic amyloidosis |
|
|
Hereditary cerebral haemorrhage with cerebral and |
|
|
systemic amyloidosis |
|
|
|
AGel |
Genetically variant gelsolin |
Autosomal dominant systemic amyloidosis |
|
|
|
|
|
Predominant cranial nerve involvement with lattice |
|
|
corneal dystrophy |
ALys |
Genetically variant lysozyme |
Autosomal dominant systemic amyloidosis |
|
|
Non-neuropathic with prominent visceral involvement |
AApoAI |
Genetically variant apolipoprotein AI |
Autosomal dominant systemic amyloidosis |
|
|
|
|
|
Predominantly non-neuropathic with prominent viscera |
|
|
involvement |
AApoAII |
Genetically variant apolipoprotein AII |
Autosomal dominant systemic amyloidosis |
|
|
Non-neuropathic with prominent renal involvement |
AApoAIV |
Apolipoprotein AIV |
Sporadic systemic amyloidosis with predominant |
|
|
cardiac and renal involvement |
|
|
|
AApoCII |
Genetically variant apolipoprotein CII |
Autosomal dominant systemic amyloidosis |
|
|
|
|
|
Non-neuropathic with prominent renal involvement |
AApoCIII |
Genetically variant apolipoprotein CIII |
Autosomal dominant systemic amyloidosis |
|
|
Non-neuropathic with prominent renal involvement |
AFib |
Genetically variant brinogen A alpha chain |
Autosomal dominant systemic amyloidosis |
|
|
Non-neuropathic with prominent renal involvement |
ALect 2 |
Leukocyte chemotactic factor 2 |
Sporadic slowly progressive renal amyloid with |
|
|
nephrotic syndrome and liver involvement |
|
|
|
ALys |
Genetically variant lysozyme |
Autosomal dominant systemic amyloidosis |
|
|
Non-neuropathic with prominent renal and hepatic |
|
|
involvement |
Diagnosis and Evaluation of Amyloidosis
As amyloidosis is a remarkably heterogeneous disease, it may present to a variety of different medical specialties. There are several reasons for patients with amyloidosis to present to a respiratory physician. Chronic pulmonary conditions can themselves give rise to systemic amyloidosis, most commonly of the AA type. Although these patients rarely present with symptomatic involvement of the lungs, the underlying pulmonary disease is the driver of the amyloidogenic protein production and it is therefore important to recognise those patients at risk. Patients with systemic amyloidosis may also present with respiratory symptoms as a consequence of the amyloidosis itself, whereby amyloid deposits are found in the lungs as a component of a more
systemic process. Localised, isolated pulmonary and respiratory tract amyloid deposits are also well-described and may either present with symptoms or may be detected incidentally on chest radiography or a biopsy [5]. Finally, it is important to recognise that, especially in the context of AL amyloidosis, pulmonary complications may also arise from treatment.
The diagnostic gold standard of amyloidosis is histological con rmation through Congo Red staining, which produces a red-green birefringence under crossed polarised light [6, 7] (Fig. 6.1). Most tissue specimens, ranging from needle biopsies to open surgical resections, can be studied, although small biopsies are open to sampling errors. A biopsy of any organ can be hazardous in amyloidosis as there is an increased risk of haemorrhage; signi cant bleeds having been reported

6 Amyloidosis and the Lungs and Airways |
79 |
|
|
a |
b |
c |
d |
Fig. 6.1 (a) Bronchial biopsy showing characteristic histological appearance of amorphous amyloid deposits stained with Congo Red. (b) The same section viewed under crossed polarised light demonstrat-
ing an apple-green birefringence. Con rmatory testing is shown by immunohistochemistry using anti-lambda light chain antibodies (c) and anti-transthyretin antibodies (d)
in 5% of liver biopsies [8], although recent data from renal biopsies have been more reassuring [9]. A case report from 1987 described fatal lung haemorrhage following a transbronchial biopsy in a patient with amyloidosis. The postmortem ndings showed that the biopsied blood vessels were in ltrated by amyloids [10]. A further case series by Utz et al. published in 1996 reported no major complications in 11 patients, following transbronchial lung biopsies; however, 2 of the 11 cases were reported to have 100 mL blood loss [11]. More encouragingly, a study from a single centre in 2017 reported no bleeding in 25 cases diagnosed by transbronchial biopsy [12]. Haemorrhage in amyloidosis is due to the increased fragility of the involved blood vessels, reduced elasticity of the amyloidotic tissues and, very occasionally, in the AL type, an acquired de ciency of clotting factors IX or X [13–15]. A less invasive alternative in suspected disease is ne needle aspiration, and this has been successfully used in the respiratory tract [16–18]. Immunohistochemical stains are then used to determine the bril protein type [5, 19].
Suitable antibodies are widely available, but, although immunohistochemistry is usually de nitive in AA amyloidosis, it is non-diagnostic in about 20% of AL deposits [20, 21]. Expertise in the typing of hereditary amyloids is restricted, and de nitive immunohistochemical typing of amyloid deposits cannot always be achieved. Mass spectrometry is extremely useful in those patients who cannot be con dently diagnosed through immunohistochemical typing; its use is limited by current availability but will become more routine in future practice [22].
If a genetic variant is suspected, then more detailed analyses examining for mutations in the gene giving rise to the amyloidogenic brils should be performed (Fig. 6.2). In general, sequencing is the preferred modality, and ideally samples should be sent to a reference laboratory with expertise in this area. A web-based repository reviewing all currently known mutations in genes with amyloidogenic potential helps guide these investigations (http://amyloidosismutations.com).
Данная книга находится в списке для перевода на русский язык сайта https://meduniver.com/
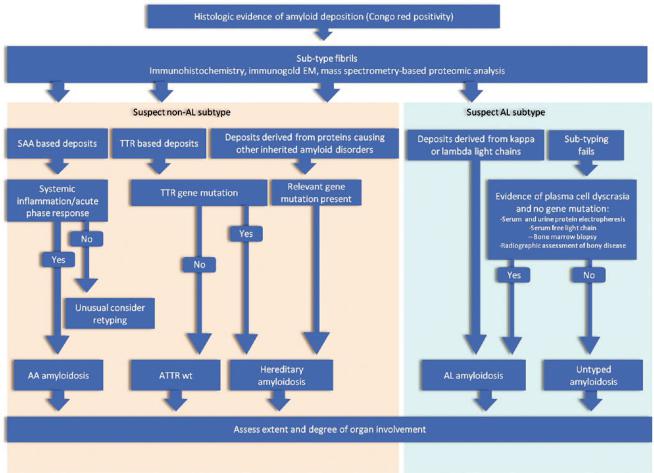
80 |
H. J. Lachmann and J. H. Pinney |
|
|
Fig. 6.2 Algorithm for investigations of patients with suspected amyloidosis
Once there is histological con rmation of amyloidosis, the extent of deposits needs to be ascertained. In respiratory tract amyloidosis, this can be challenging and the optimum imaging technique can vary depending upon the distribution of deposits. Plain radiography as an initial assessment can be helpful but may be normal in many cases. Computed tomography (CT) scanning is particularly useful in further de ning interstitial diseases. In combination with positron emission tomography (PET) imaging, it can also help to better de ne the metabolic activity of a solid lesion, thus aiding in differentiation from more typical intrathoracic malignancies or metastases as well as rare entities such as plasmacytomas [23]. In addition, magnetic resonance imaging (MRI) and bronchoscopy may also be useful in combination with comprehensive pulmonary function tests (PFTs). PFTs are an important objective tool to formally establish the severity of clinically relevant diseases and are useful in guiding therapeutic decisions [5, 24]. Evidence of systemic diseases should be sought clinically by performing haematological and biochemical pro les. The plasma cell clones that underlie systemic AL amyloidosis are often subtle and may not be
detected by bone marrow examination or immuno xation of the serum and urine; use of a serum-free light-chain assay increases diagnostic sensitivity [25–28]. Immunoglobulin gene rearrangement studies may identify subtle clones in either the bone marrow or, in the case of localised AL, within the amyloid biopsy material [29] (Fig. 6.1c, d).
Radiolabelled SAP speci cally localises to amyloid deposits in vivo in proportion to the quantity of amyloids present and thereby enables diagnosis, quanti cation and monitoring of the amyloids [2]. SAP scintigraphy is useful in visualising amyloids in solid organs; localisation to the lungs is poor and not routinely used for monitoring amyloid deposits in the respiratory tract (Fig. 6.3a, b). Cardiac amyloidosis is best evaluated by a combination of echocardiography, electrocardiogram (ECG) and cardiac magnetic resonance imaging (CMR). Two-dimensional Doppler echocardiography classically reveals concentric biventricular wall thickening with a restrictive lling pattern [30] (Fig. 6.3b). Amyloidosis causes diastolic dysfunction with preserved contractility until an extremely late stage [31]. The ECG may be normal in patients with substantial cardiac

6 Amyloidosis and the Lungs and Airways |
81 |
|
|
Fig. 6.3 Scintigraphic assessments using 123I-human SAP. An anterior whole body scintigraphic image from a patient obtained following intravenous injection of 123I-human SAP showing abnormal uptake into the amyloid deposits within the spleen, liver and bone marrow (a). An anterior plasmacytoma and deposition in the spleen. An anterior whole body scintigraphic image from a patient with a solitary intrathoracic amyloidoma (b) with corresponding SPECT-CT (c)
a |
b |
c
amyloidosis, but, in advanced disease, it commonly shows small voltages, pathological ‘Q’ waves (a pseudo-infarct pattern) in the anterior chest leads and conduction abnormalities. CMR is extremely useful in identifying cardiac amyloidosis. Typical appearances are of homogeneous late gadolinium enhancement [32]. 99mTc-3,3-diphosphono-1,2- propanodicarboxylic acid (99mTc-DPD) scintigraphy is a speci c test indicative of bril deposition within the heart, with data suggesting that the degree of uptake and pattern of distribution in the extracardiac soft tissue may be speci c for amyloid transthyretin (ATTR) amyloidosis [33, 34].
Elevation of N-terminal pro-brain natriuretic peptide (NT-Pro-BNP) and cardiac troponins can also be helpful in establishing whether a patient has cardiac amyloidosis [35, 36]. These enzymes are not speci c and can be elevated for other reasons such as renal impairment and other forms of cardiomyopathy; however, a normal NT-Pro-BNP can exclude cardiac involvement [37].
The key to effective monitoring of amyloidosis and its treatment is relatively frequent repetition of these investigations, bearing in mind that organ dysfunction may not closely refect amyloid load.
Данная книга находится в списке для перевода на русский язык сайта https://meduniver.com/