

3.13. Константы скоростей химических реакций
Успешная работа таких программ по предсказанию химических процессов требует знания констант скоростей. Теоретические методы их определения пока работают лишь для простейших систем. Для обработки и систематизации экспериментальных данных (основного источника k) до сих пор используется эмпирическое уравнение Аррениуса
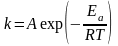 (3.62)
(3.62)
где Ea – энергия активации, A – предэкспоненциальный множитель.
В нижеследующей табличке представлены примеры экспериментально найденных кинетических констант.
Уравнение реакции |
Среда |
Порядок реакции |
A |
Eа, кДж/моль |
C2H5Br = C2H4 + HBr |
газ |
1 |
7,210–12 c–1 |
218,0 |
H2 + C2H4 = C2H6 |
газ |
2 |
4107 м3/(мольc) |
180,5 |
CH3Br + J– = CH3J + Br– |
р-р H2O |
2 |
1,7107 м3/(мольc) |
76,6 |
3.14. Теория (модель) активированного комплекса
Углубленное представление об элементарном акте химических реакций дает теория или модель активированного комплекса (МАК). При этом предполагается, что при сближении реагирующих молекул наблюдается почти непрерывный переход из начального в конечное состояние, который протекает через переходное состояние или активированный комплекс (АК) с максимумом потенциальной энергии Епот . Разность Епот активированного комплекса и исходных веществ, своеобразный энергетический барьер, названа энергией активации Еа реакции.
Еа определяется по Епот как функции взаимных расстояний всех участвующих в реакции атомов. Для линейного расположения атомов А, В и С простой реакции: А + ВС → (А∙В∙С)→АВ + С результат квантово-химического расчёта Епот от межъядерных расстояний
δ
Еа
Потенциальная
энергия
Пространственная
координата реакции
|
|
Рис.3.3. Потенциальная энергия вдоль координаты реакции |





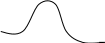






 хB
– C и хA
- B можно
наглядно
отобразить
в виде диаграммы потенциальных кривых.
Наиболее вероятный путь реакции (при
наименьшей затрате энергии) ведет
через состояние активированного
комплекса. Изменение Епот
вдоль
координаты реакции схематично показано
на рис.3.3
хB
– C и хA
- B можно
наглядно
отобразить
в виде диаграммы потенциальных кривых.
Наиболее вероятный путь реакции (при
наименьшей затрате энергии) ведет
через состояние активированного
комплекса. Изменение Епот
вдоль
координаты реакции схематично показано
на рис.3.3
Обозначим стехиометрические коэффициенты реагентов 'i ai, а переходное состояние активированного комплекса A, тогда химическое уравнение однобарьерной реакции
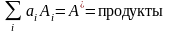 .
(3.63)
.
(3.63)
Определяющий постулат МАК (Эйринг, Поляни):
"Скорость реакции равна концентрации c активированного комплекса на вершине барьера, помноженной на частоту ν пересечения этого барьера."
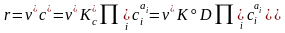 .
(3.64)
.
(3.64)
В
(3.64) использованы: записанный для
реагентов и АК «концентрационный» ТЗДМ
 ,
взаимосвязь констант
,
взаимосвязь констант
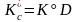 (см. (3.50с))
и
введено обозначение D
= (RT/Р°)-Δν
= (RT/P°)n-1,
(см. (3.50с))
и
введено обозначение D
= (RT/Р°)-Δν
= (RT/P°)n-1,
а Δν=1 - n выражено через порядок реакции n.
Из сопоставления (3.64) с КЗДМ (3.58) константа скорости равна
k = ν≠K° D. (3.65)
Активированный комплекс (АК) как неустойчивое переходное образование, в котором прежние химические связи еще не разорваны, а новые еще не до конца образовались, характеризуется избытком колебательных степеней свободы. При этом из константы равновесия K° можно выделить колебательную составляющую, определяющую переход через АК: K° = K≠ exp(–ΔG( ν≠)/RT ) ≈ K≠∙kБT/(h ν≠).
Подстановка полученного выражения в (3.65) дает окончательную формулу для константы скорости в МАК:
k = (kБT/h) D K≠ = (kБT/h) D exp(-ΔrG≠/RT). (3.65')
k = (kБT/h) D exp( -ΔrH≠/RT + ΔrS≠/R ) (3.65'')
Сопоставление (3.65'') с формулой Аррениуса раскрывает смысл эмпирически введенных энергии активации, как энтальпии реакции перехода реагентов в АК, а также предэкспоненциального сомножителя, образованного универсальным частотным фактором и энтропией реакции перехода реагентов в АК.
Ea= ΔrH≠ , A=(kБT/h) D exp(ΔrS≠/R). (3.66)
Размерность константы скорости определяется размерностями входящих в А частотного фактора [kБT/h] = 1/с и [D] = [К≠с].
Главное в том, что теоретическое выражение МАК определяет квантово-статистический способ расчета констант скоростей химических реакций. А самое замечательное при выводе − это сокращение постулируемой частоты пересечения барьера и выделенной из АК частоты слабейшего колебательного звена, определяющего реакционное преобразование компонентов.