

u_sem
.pdf30
Если a и b начальные концентрации А и В, а x – текущая концентрация С или D, тогда выражение для константы скорости реакции второго порядка примет вид:
k = |
1 |
ln |
[b(a-x)] |
|
|
|
|||
t(a-b) |
[a(b-x)] |
|||
|
|
По типу реакций второго порядка протекает большое число гомогенных реакций в растворе, например, реакции димеризации или реакции нуклеофильного замещения второго порядка.
Реакции более высоких порядков чрезвычайно редки, так как вероятность столкновения трёх или более частиц очень мала.
4.3. Понятие о лимитирующей стадии реакции.
Большинство органических реакций представляет собой совокупность последовательных стадий, приводящих исходные реагенты к продукту реакции. Каждая из этих стадий протекает со своей скоростью. Следовательно, существует такая стадия, которая протекает с наименьшей по сравнению с другими стадиями этого процесса скоростью. Эта стадия называется лимитирующей стадией. Лимитирующая стадия определяет общую скорость реакции. Многостадийные процессы имеют аналогию с набором воронок с различным диаметром трубок вставленных одна в другую, через которые течёт вода. Воронка с самой узкой трубкой определит скорость протекания воды.
Скорость химической реакции выражается уравнением, которое связывает константу скорости реакции k с температурой Т. Это соотношение известно как уравнение Аррениуса.
lg k = lg А ─ ЕА /2,303RT,
где А – предэкспоненциальный множитель (частотный фактор); ЕА – энергия активации; R – газовая постоянная; Т – температура (К).
Из уравнения Аррениуса следует, что чем выше температура, тем больше константа скорости. Если построить график зависимости lg k от 1/Т,
то получится прямая, угол наклона которой равен ЕА / 119,147 (Дж·К-1·моль-1) Из представленной зависимости следует ещё один вывод: чем больше
энергия активации реакции, тем быстрее происходит изменение скорости реакции с изменением температуры
Энергия активации. Минимальное количество энергии, которое получают реагирующие молекулы или частицы при столкновении и которое необходимо для осуществления реакции, называется энергией активации. Для химического взаимодействия необходимо, чтобы реагирующая система обладала энергией, по крайней мере, равной энергии активации данной реакции.
31
Из уравнения Аррениуса следует, что скорость реакции, как мера реакционной способности соединения определяется тремя факторами: частотой столкновений реагирующих соединений, фактором вероятности и энергетическим фактором. Если температура и концентрация в ряду родственных реакций постоянна, то практически постоянно число столкновений. Следовательно, в ряду близких по строению соединений остаётся практически постоянным и фактор вероятности, а основной вклад в различную реакционную способность вносит энергетический фактор – энергия активации. Она и опре-
деляет относительную реакционную способность соединений.
4.4. Тепловой эффект реакции.
Для процессов, протекающих при постоянном давлении (сюда относится большинство реакций, проводимых в лаборатории), теплота реакции равна изменению энтальпии ( Н) в ходе реакции. В свою очередь изменение энтальпии, сопровождающее реакцию, равно теплоте образования продуктов минус теплота образования исходных веществ:
Н = ∑ Нf продуктов – ∑ Нf исходных веществ
Если в ходе реакции выделяется тепло, то реакция называется экзо-
термической (отрицательное значение Н). Реакция, сопровождающаяся поглощением тепла, называется эндотермической (положительное значение Н), например, при хлорировании метана, протекающая по уравнению:
CH4 + Cl2 → CH3─Cl + HCl,
разрываются две связи (С─Н и Cl─Cl), на что требуется (427 + 242,7) = 669,7 кДж/моль. Образуются две связи С ─Cl и Н─ Cl и при этом выделяется 770,2 (339,1 + 431,1) кДж/моль, т.е. в результате реакции выделяется 100,5 (770,2 – 669,7) кДж/моль тепла. Н = –100,5 кДж/моль.
4.5. Переходное состояние.
Химическая реакция представляет собой непрерывный процесс, заключающийся в постепенном переходе от исходных веществ к конечным. Во многих химических реакциях реагирующая система преодолевает энергетический барьер, высота которого определяется потенциальной энергией исходных соединений и энергией активации данной реакции
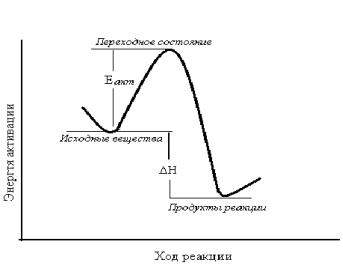
32
Состояние реагирующей системы в момент, когда она обладает максимальной энергией (вершина энергетической диаграммы) называется переход-
ным состоянием.
Принято считать, что переходное состояние – это особое состояние реагирующих веществ, которое нельзя изучить экспериментально с целью определения его структуры, так как время жизни переходного состояния соответствует времени единичного колебания химической связи (около 10-13 с).
Рис. 4.1 Изменение потенциальной энергии в процессе реакции: энергия переходного состояния соответствует максимуму на кривой.
Любые факторы, способствующие уменьшению энергии переходного состояния, ускоряют его образование и тем самым ускоряют реакцию. По-
этому при рассмотрении влияния различных факторов на скорость реакции (природы растворителя, катализатора или структуры реагентов) необходимо провести анализ структуры переходного состояния и факторов, влияющих на его устойчивость.
Информацию о структуре переходного состояния получают косвенными методами. Один из них заключается в рассмотрении устойчивости тех промежуточных частиц (интермедиатов), которые образуются в ходе реакции. Например, в реакции электрофильного замещения в ароматическом кольце моделью переходного состояния может служить σ-комплекс.
Другой подход к интерпретации геометрии переходного состояния является использования постулата Дж. С. Хеммонда, в соответствии с которым переходное состояние:
а) для сильно экзотермических реакций с малой энергией активации по своему строению близко к исходным реагентам,
б) для сильно эндотермических реакций сходно со структурой продуктов реакции,

33
в) для реакций с незначительным тепловым эффектом и большой энергией активации отличается по строению, как от исходных реагентов, так и от продуктов реакции.
4.6.Катализ и скорость реакции.
Вприсутствии катализатора скорость реакции увеличивается, так как
катализатор направляет реакцию по новому пути с более низким значением энергии активации (через переходное состояние с меньшей энергией). Например, гидрирование алкенов – экзотермическая реакция (выделяется приблизительно 125,6 кДж/моль тепла), но в отсутствие катализатора она протекает с незначительной скоростью, даже при повышенной температуре, следовательно, в отсутствие катализатора реакция гидрирования алкенов имеет большую энергию активации. На практике реакцию гидрирования проводят, используя в качестве катализаторов металлы платиновой группы (Pt, Pd, Rh) или никель Ренея. Эти металлы нерастворимы в органических растворителях
икатализ ими осуществляется гетерогенно.
Реагенты (алкен и водород) адсорбируются на огромной поверхности тонко измельчённого металла, где и протекает реакция. Реакция адсорбированных молекул сильно отличается от реакции обычных молекул; предполагают, что на поверхности катализатора сначала происходит разрушение π- связи алкена, а затем уже присоединение, адсорбированного поверхностью катализатора водорода.
Рис. 4.2. Влияние катализатора на изменение потенциальной энергии реагирующей системы: присутствие катализатора ( ▬▬ ), отсутствие катализатора (- - - - ).
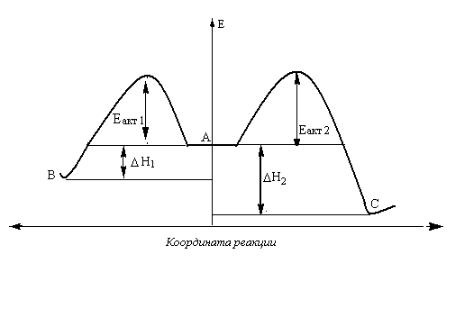
34
Использование катализаторов в реакции гидрирования алкенов позволяет проводить процесс при комнатной температуре и избыточном давлении водорода равном 1 атмосфере.
4.7. Термодинамический и кинетический контроль реакции
Существует много органических реакций, в которых энергетические требования для нескольких направлений реакции сходны. Если реагирующая система может одновременно изменяться по двум или более конкурирующим направлениям, то важно уметь проанализировать факты, которые могут сделать одно направление доминирующим.
Термины кинетический и термодинамический контроль особенно по-
лезны при обсуждении случаев, в которых путём изменения условий реакции можно смещать состав продукта от ожидаемого при кинетическом контроле к составу продукта, обусловленного термодинамическим контролем.
Если соотношение продуктов реакции определяется скоростями их образования, то такие реакции называются кинетически контролируемыми.
Если исходное вещество и один из продуктов реакции находятся в состоянии равновесия, то состав продуктов реакции может определяться тер-
модинамикой равновесной системы. Если это так, то говорят, что состав продукта регулируется термодинамическим контролем. На рис. 4.3 приведе-
на энергетическая диаграмма для реакции:
|
|
k1 |
k2 |
|
||
B |
|
|
|
A |
C |
|
|
|
|
|
|||
|
|
|
|
|||
|
|
k-1 |
|
|
||
Рис.4.3. Энергетическая диаграмма
Если продукт С термодинамически более устойчив, чем А и В, но энергия активации превращения А в В меньше, чем А в С (Еа1 < Еа2 ), тогда можно
35
подобрать такие внешние условия проведения реакции, когда не преодолевается энергетический барьер Еа2 тогда конечным продуктом будет преимущественно продукт В (кинетически контролируемый). При увеличении продолжительности реакции, или если условия реакции позволяют преодолеть барьер Еа2 будет образовываться термодинамически более стабильный продукт С
(термодинамически контролируемый). Продукт В будет также превращаться в А и затем в С. Например, при электрофильном присоединении брома к бу- тадиену-1,3 может образоваться 1,2-дибромбутен-3 (продукт 1,2- присоединения) и 1,4-дибромбутен-2 (продукт 1,4-присоединения). Соотношение между образующимися продуктами в этой реакции определяется как кинетикой реакции, так и термодинамической устойчивостью образующихся продуктов. В случае присоединения брома к бутадиену-1,3 при низкой температуре (-80 ºС) образуется 80% 1,2-дибромбутена-3 (кинетически контролируемый продукт). При температуре + 40 ºС преодолевается энергетический барьер на пути образования продукта 1,4-присоединения, кроме того увеличивается скорость превращения продукта 1,2-присоединения в исходный бу- тадиен-1,3, поэтому образуется 80% 1,4-дибромбутен-2 (термодинамически контролируемый продукт).
5. ХАРАКТЕРИСТИКА ИНТЕРМЕДИАТОВ РЕАКЦИЙ
5.1. Интермедиаты в многостадийных реакциях
Описание интермедиатов в многостадийных реакциях является основной целью исследования их механизмов. Если достаточно хорошо понята природа каждого интермедиата в реакции, то механизм реакции можно считать в значительной мере выясненным. Количество интермедиата в любой момент времени зависит от скорости стадии, в которой он образуется и от скорости стадии его последующего превращения:
к1 к2
реагенты → интермедиат → продукты реакции
Для многих реакций к1 >> к2 и интермедиат можно действительно выделить. Любой истинный интермедиат, выделенный из прерванной реакции, помещённый снова в условия реакции должен давать ожидаемый продукт реакции. Если скорость образования интермедиата только немного превышает скорость его исчезновения, то концентрация интермедиата в любой момент времени невелика и его не удаётся выделить. Иногда интермедиат удаётся «поймать в ловушку»: к реакционной системе добавляют соединение, которое должно специфически реагировать с интермедиатом. При этом интермедиат выключается из нормального процесса, а доказательство его существования можно получить, установив строение продукта захвата. Так, предположение
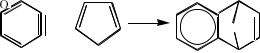
36
об образовании дегидробензола в некоторых реакциях металлорганических соединений удалось подтвердить, уловив дегидробензол в виде аддукта с фураном:
+ |
O |
Более эффективным для установления природы интермедиатов, присутствующих в реакционной среде в низких концентрациях является применение инструментальных методов.
Применение электронной спектроскопии в ультрафиолетовой и видимой области имеет самую давнюю историю. Современные приборы позволяют быстро пробегать ультрафиолетовую и видимую области спектра; полученные спектры могут дать определённые доказательства образования промежуточных частиц. В благоприятных случаях интермедиат может быть обнаружен в концентрации ниже 10 5 М. Ограничением в применения электронной спектроскопии является то, что разыскиваемый интермедиат должен иметь поглощение в области 220 – 700 нм.
Инфракрасные (ИК) спектрометры измеряют поглощение энергии при возбуждении молекулярных колебаний, включая валентные, деформационные и крутильные колебания различных частей молекулы. Если определённая функциональная группа, имеющая характеристическое поглощение, присутствует в интермедиате, то при изучении изменений спектра в характеристической области, отвечающей поглощению этой функциональной группы можно обнаружить интермедиат. Например, если интермедиатом может быть кетен, характеристическое поглощение, которого лежит в области 2100 – 2130 см 1, то обнаружение этого характеристического поглощения при ИКспектроскопическом контроле протекания реакции является хорошим доказательством кетеновой природы интермедиата. В некоторых случаях методом ИК-спектроскопии удаётся зафиксировать интермедиат в концентрации порядка 10 3 М.
Очень широко используется спектроскопия ядерного магнитного резонанса. Принципиальным ограничением метода является не очень высокая чувствительность метода.
Свободные радикалы можно обнаружить в чрезвычайно низких концентрациях с помощью электронного парамагнитного резонанса (ЭПР). Диамагнитные молекулы, присутствующие в растворе, не дают сигнала, и, следовательно, возможности помех сильно уменьшены. В некоторых случаях удаётся обнаружить радикальные частицы в диапазоне 10 6 − 10 8 М.
В последние годы наряду с методом ЭПР для изучения механизмов реакций и фиксации свободнорадикальных стадий процесса широко применяется метод химической поляризации ядер (ХПЯ). Смысл явления ХПЯ заключа-
37
ется в том, что при проведении химической реакции в магнитном поле в тех случаях, когда реакция идет с промежуточным образованием свободных радикалов, в спектрах магнитного резонанса продуктов может обнаруживаться или аномально большое поглощение, или радиоизлучение, которое может быть зафиксировано в течение времени ядерной релаксации. Наличие ХПЯ в продукте может служить признаком того, что он образовался в результате рекомбинации свободнорадикальной пары, а вид спектра дает возможность судить о природе этой пары. Использование ХПЯ позволило подтвердить свободнорадикальный характер некоторых перегруппировок, а также сделать вывод о механизме распада азосоединений. С помощью ХПЯ удается различить, проходят реакции карбенов через их синглетное или триплетное состояние. В ряде случаев метод ХПЯ позволяет не только сделать качественные выводы о механизме процесса, но и оценить скорости быстрых элементарных стадий.
Следует отметить, что регистрация в реакционной смеси малостабильной частицы не всегда может считаться однозначным доказательством того, что основная реакция идёт именно через неё. Иногда малостабильные промежуточные частицы не сопровождают основной путь реакции, а получаются в побочном процессе. Например, в реакции хлорирования гексаметилбензола был предложен следующий механизм:
|
CH |
|
|
|
CH3 |
|
|
|
|
|
|
|
|
CH |
||||||||||||||||||||||
H3C |
|
|
3 |
|
|
|
|
|
|
|
|
H3C |
|
|
|
|
|
CH3 |
|
|
|
|
|
|
|
|
|
2 |
|
|
|
|||||
|
|
|
|
CH |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
H C |
|
|
|
|
CH |
|||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||||||||
|
|
|
|
|
3 |
|
|
Cl |
|
|
|
|
|
|
|
|
|
3 |
|
|
|
|
3 |
|
|
|
||||||||||
|
|
|
|
|
|
|
|
|
|
H3C |
|
|
|
|
|
CH3 |
|
|
Cl |
|
|
|
|
|
|
|
|
|
|
|
Cl |
|||||
H3C |
|
|
|
|
CH |
|
|
|
|
|
|
|
|
|
|
|
H |
|
|
H C |
|
|
|
|
CH |
|||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||||||||||||
|
|
3 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
3 |
|
3 |
|
|
|
|||||||||||||
|
CH |
|
|
|
CH3 |
|
|
|
|
|
|
|
|
CH |
||||||||||||||||||||||
|
3 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
3 |
|
|
|
|
||||||
Этот механизм считался доказанным, так как катион-радикал был зафиксирован в реакционной смеси. Впоследствии появились доказательства того, что реакция идёт как электрофильное замещение, а катион-радикал образуется в результате побочного процесса.
|
CH3 |
|
|
|
|
|
|
|
CH3 |
|
|
|
|
|
|
CH2Cl |
|||||||||||||
H3C |
|
|
|
CH |
|
|
|
|
|
|
H3C |
|
|
|
|
|
|
CH |
|
|
|
|
|
H C |
|
|
|
CH |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
3 |
|
|
|
|
|
|
|
|
|||||||
|
|
|
|
3 |
|
|
|
Cl |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
3 |
|
|
|
3 |
|
H3C |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
H |
|
|
|
|
|
|
|
|
||
|
|
|
CH3 |
|
|
|
|
|
|
H3C |
|
|
|
|
|
|
|
|
|
|
|
H3C |
|
|
|
CH |
|||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
CH |
|
|
|
|
|
|
|
|||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
3 |
|||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
3 |
|
|
|
|
|
|
|
|
|
|
|||||
|
CH3 |
|
|
|
|
|
|
CH |
3 |
|
|
Cl |
|
|
|
|
|
|
CH |
||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
3 |
|
|||
В органической химии известно пять типов частиц, в которых валентности атомов углерода равны двум или трём. Это карбокатионы (1), карбанионы (3), свободные радикалы (2), карбены (4) и арины (5):

38
|
|
R |
|
|
|
|
|
|
R |
|
|
R |
|
|
R |
|
|
|
R |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
C |
|
|
|
R |
|
C |
R C |
|
C |
|
||||||
|
|
R |
|
|
|
|
|
R |
|
|
R |
|
R |
|
||||
|
|
|
|
|
|
|
|
|
|
|
||||||||
|
1 |
|
2 |
3 |
4 |
5 |
||||||||||||
5.2. Карбокатионы.
Органические катионы, содержащие положительно заряженный атом углерода, могут быть двух типов: карбкатионы с трёхвалентным атомом углерода R3C+ и ониевые катионы, образующиеся при присоединении протона к насыщенному атому углерода.
Представление о природе органических катионов первого типа появилось в результате исследований производных трифенилметана. При измерении электропроводности растворов трифенилметилгалогенидов в жидком сернистом ангидриде было обнаружено, что они ведут себя как ионные соединения. Для раствора трифенилкарбинола в серной кислоте была предложена структура соли:
(C6H5)3C–OH + H2SO4 → (C6H5)3C+HSO4 + Н2О
Ионный характер трифенилметилперхлората −(C6H5)3C+ClO4 убедительно доказан рентгеноструктурным анализом.
Было высказано предположение, что карбокатионы могут возникать как быстро превращающиеся промежуточные продукты при самых различных органических реакциях.
В карбокатионах положительно заряженный атом углерода имеет на внешней оболочке шесть электронов и, следовательно, находится в sp2- состоянии. Все три атома, соединённые с ним σ-связями, расположены в одной плоскости в углах правильного треугольника. Вакантная р-орбиталь перпендикулярна плоскости треугольника:
Рис. 5.1. Строение карбокатиона: вакантная р-орбиталь перпендикулярна плоскости σ-связей.
39
Согласно квантовомеханическим расчётам плоская конфигурация карбокатиона стабильнее пирамидальной конфигурации на 84 кДж/моль.
Многочисленные исследования, проведённые с целью выяснения влияния структуры на устойчивость карбокатионов, показали, что любое влияние,
делокализующее положительный заряд карбокатиона ведёт к стабилизации,
что означает понижение энергии основного состояния. Следовательно, наличие электронодонорных заместителей в структуре карбокатиона стабилизирует, а электроноакцепторных — дестабилизирует его.
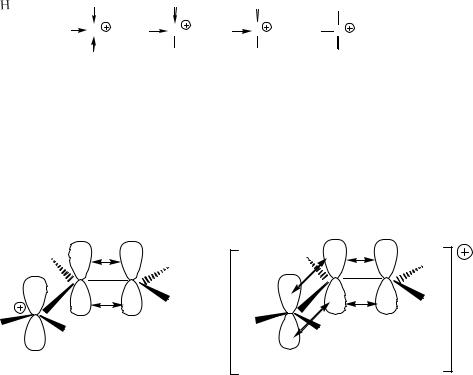
Стабилизация карбокатионов за счёт явления гиперконъюгации.
Известно, что алкильные группы по сравнению с водородом стабилизируют карбокатионы.
Высказано предположение, что стабилизация осуществляется за счёт делокализации электронов С-Н связей этих групп при участии вакантной р-орбитали
(гиперконъюгация).
Отсюда в ряду простых алкильных катионов устойчивость возрастает в ряду: первичные, вторичные, третичные:
R |
R |
|
H |
R C |
R C |
R C |
H C |
R |
H |
H |
H |
Стабилизация карбокатионов за счёт эффекта сопряжения. Расчёты показывают, что аллильный карбокатион (СН2=СН–СН2+) устойчивее метильного на 250,5 кДж/моль, а бензильный карбокатион (С6Н5СН2+ ) — на 314 кДж/моль. Это связано с делокализацией π -электронов двойной связи в случае аллильного катиона и π-электронов бензольного кольца в случае бензильного катиона:
|
H |
H |
|
H |
H |
|
|
|
|
||
|
C |
C |
|
C |
C |
|
C |
H |
|
C |
H |
H |
|
H |
|
||
|
H |
|
|||
|
H |
|
|
|
Рис. 5.3. Стабилизация аллильного катиона за счёт эффекта сопряжения.
На рис. 5.3 для сравнения показан локализованный катион, который реально не существует. Поскольку положительный заряд распределён между двумя атомами углерода. Реакции, в которых возникают аллильные карбокатионы, часто сопровождаются перегруппировками аллильной системы. На-