

u_sem
.pdf100
2Fe + 3Cl2 = 2FeCl3; FeCl3 + Cl2 = FeCl4 + Cl+
AlCl3 + Br2 = AlCl3Br + Br+
Галогенирование активных аренов свободными галогенами протекает очень легко и часто приводит к полигалогенопроизводным. Например, анилин и фенол при действии бромной воды сразу превращаются в нерастворимые 2,4,6-трибромпроизводные. Фенол при избытке реагента превращается в 2,4,4,6 тетрабромциклогексадиен-2,5-он:
OH |
|
|
|
OH |
|
|
|
|
|
O |
|||||||||||||
|
|
|
|
|
|
|
Br2 , H2O, 20 |
o |
C |
Br |
|
|
|
Br |
Br2 , H2O, 20 o C |
Br |
|
|
|
|
|
Br |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Br Br |
||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||
|
|
|
|
|
|
|
|
|
|
|
Br |
|
|
|
|
|
|||||||
Иодирование. Из всех галогенов иод обладает самой низкой электрофильностью. Для его активации применяют окислители или кислоты Льюиса. Нередко иодирование осуществляют соединениями, содержащими связанный электроположительный иод. В качестве окислителей используют азотную или иодную кислоты, перманганат калия, перекись водорода и др. Роль окислителя заключается в окислении I2 до 2I+. Классическим примером введения иода с использованием окислителя является иодирование с использованием системы I2 − HNO3 – H2SO4 в ледяной уксусной кислоте. Нитробензол этой системой иодируется, образуя 3-иоднитробензол с выходом 61%.
Алкилирование по Фриделю – Крафтсу. Реакция Ш. Фриделя и Дж.
Крафтса (1877 г.) представляет собой удобный метод прямого введения алкильной группы в ароматическое кольцо. Алкилирование ароматических соединений осуществляется под действием алкилгалогенидов в присутствии катализаторов – кислот Льюиса. Наиболее активными катализаторами являются безводные сублимированные бромиды алюминия и галлия. Очень часто используется AlCl3. Согласно одному из предложенных механизмов электрофилом служит карбокатион, образующийся при реакции кислоты Льюиса с алкилгалогенидом:
RCl + AlCl3 = AlCl4 + R+
R+ + C6H6 → C6H6+−R медленно
C6H6+−R + AlCl4 → С6H5R + HCl + AlCl3 быстро
В другом механизме электрофилом служит алкильная группа полярного комплекса AlCl3 и алкилгалогенида:
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
101 |
||||
|
|
Cl |
|
|
|
|
|
|
|
|
|
R |
|
|
|
|
|
||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||||
Cl |
|
|
Al |
|
|
|
Cl |
|
R |
|
|
|
C6H6 |
|
C6H5 |
H |
|
|
AlCl4 |
||||
|
|
|
|
|
|
|
|
|
|
|
|
||||||||||||
|
|
|
|
|
|
|
|
|
|||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
||||||||||||
|
|
|
|
|
|
|
|
|
|
||||||||||||||
|
Cl |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||
Точное строение интермедиата не известно. |
Полагают, что строение |
||||||||||||||||||||||
комплексов RX•MXn постепенно изменяется от структуры координационного аддукта в случае R= CH3 до структуры ионной пары в случае R= t-Bu. Однако
экспериментально это пока не подтверждено.
Способность атома галогена в RX к комплексообразованию с кислотой Льюиса резко уменьшается от фтора к иоду, вследствие этого активность алкилгалогенидов в качестве алкилирующих агентов в реакции Фриделя - Крафтса также уменьшается в ряду RF >RCl > RBr > RI. По этой причине алкилиодиды не применяют в качестве алкилирующих реагентов.
Ароматические углеводороды легко алкилируются самыми разнообразными алкилгалогенидами, аллилгалогенидами, бензилгалогенидами и триарилметилгалогенидами в присутствии FeCl3 , FeBr3 , AlCl3 , AlBr3 при 0 – 25 ºС или при нагревании. В качестве алкилирующих реагентов также используют алкены или спирты в присутствии BF3 или протонных кислот (фосфорной, полифосфорной или серной):
|
H |
R |
H2O |
R-OH H |
R-O H |
Наиболее дешевыми реагентами для проведения алкилирования в промышленности являются алкены – этилен, пропилен, изобутилен и др. Реакция алкилирования бензола алкенами лежит в основе крупнотоннажного получения этилбензола и изопропилбензола (кумола).
Алкилирование можно проводить внутримолекулярно. Так 4-фенил-1- бутанол в фосфорной кислоте даёт продукт внутримолекулярной циклизации:
CH2 |
CH2 |
|
H3PO4 |
|
|
|
|
CH2 |
CH2 |
|
|
|
|
|
|
|
|
|
CH2 |
CH2 |
|||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||||
|
|
|
H2O |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
H |
|
|
|
|
|
|
|
|
|
||
|
CH2 |
|
|
|
|
|
|
|
|
CH2 |
|
|
|
|
|
|
|
CH2 |
CH2 |
||||||||
CH2 |
|
|
|
|
|
|
CH2 |
|
|
|
|
|
|
|
|
||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||
OH
Алкилирование аренов как синтетический метод имеет ряд серьёзных недостатков, ограничивающих его применение. Во-первых, образующийся продукт алкилирования более реакционноспособен, чем исходный арен. Поэтому алкилирование аренов при соотношении реагентов близком к эквимолярному приводит к образованию значительного количества продуктов полиалкилирования. Для того чтобы свести полиалкилирование к минимуму, необходимо использовать большой избыток ароматического углеводорода, что
102
не всегда можно осуществить. Во-вторых, в процессе алкилирования происходит изомеризация алкилирующего реагента. Так, при алкилировании бензола 1-хлорпропаном образуется смесь пропилбензола и изопропилбензола, в которой больше изомеризованного продукта:
CH3CH2CH2Cl |
AlCl3 |
|
|
CH2CH2CH3 |
|
|
CH(CH3)2 |
||||||
|
|
|
|||||||||||
|
|
|
|
|
|
|
|
|
|
||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||
35% |
|
|
|
65% |
|
||||||||
В-третьих, в ходе алкилирования по Фриделю – Крафтсу происходит миграция алкильных групп в конечном продукте реакции:
|
AlCl |
|
|
|
CH(CH3)2 |
|
|
CH3 |
|
|
|
CH(CH ) |
||
|
|
|
|
|
|
|
|
|||||||
|
|
|
|
|
|
CH |
|
|
|
|
|
|||
|
3 |
|
|
|
|
|
|
|
|
|
|
|
3 2 |
|
(CH3)2CHCl |
|
|
|
|
|
|
3 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
CH3 |
|
|
|
|
|
|
|
|||||
|
|
|
|
|
|
|
|
|
||||||
|
|
|
|
|
|
|
|
|
|
|
CH3 |
|||
Ароматические соединения, содержащие электроноакцепторные замес-
тители NO2 , NO, CN, COOR и др. не алкилируются в условиях реакции Фри-
деля – Крафтса. Ароматические фенолы или амины связывают кислоты Льюиса в нереакционноспособный донорно-акцепторный комплекс.
Ацилирование по Фриделю – Крафтсу. Реакция нашла широкое при-
менение для получения жирно-ароматических кетонов:
O |
C R |
R C Cl |
O |
Ацилирующими агентами являются галогенангидриды и ангидриды кислот в присутствии кислот Льюиса − галогенидов алюминия, трифторида бора или пентафторида сурьмы. Ацилгалогениды и ангидриды кислот образуют с кислотой Льюиса донорно-акцепторные комплексы состава 1:1 и 1:2. Спектральными методами было установлено, что кислоты Льюиса координируются по карбонильному атому кислорода ацилгалогенида или ангидрида кислоты.
Образующийся алкилароматический кетон представляет собой более сильное основание Льюиса, чем ацилгалогенид, даёт стабильный комплекс с кислотой Льюиса, поэтому для ацилирования ароматических соединений ацилгалогенидами требуется больше одного моля катализатора на моль арена. Кетон выделяют, разлагая его комплекс с кислотой Льюиса водой или соля-
103
ной кислотой. Жирноароматические кетоны получают, как правило, с высоким выходом, так как реакция ацилирования лишена тех недостатков, которые присущи реакции алкилирования. Так, например, ацетилирование нафталина хлористым ацетилом в сероуглероде в присутствии AlCl3 даёт метил-1- нафтилкетон с выходом 92%.
Ориентация входящей ацильной группы зависит от её природы. Хлорангидриды и ангидриды алифатических карбоновых кислот вступают в основном в n-положение, если ароматический субстрат содержит заместитель первого рода. Соотношение орто/пара - изомеров не превышает 0,03. Содержание метаизомера не превышает 0,5%. Таким образом, ацилирование аро-
матических соединений хлорангидридами жирных кислот осуществляется чрезвычайно региоселективно в параположение. Ацилирование хлорангидри-
дами ароматических кислот не является столь селективной реакцией (возрастает доля ортоизомера). Это объясняют меньшей долей стерических препятствий, так как катион ацилия (ArCO+) образующийся в результате взаимодействия галогенангидрида ароматической кислоты с кислотой Льюиса, имеет меньший объём, чем комплекс AlkCOCl AlCl3:
C |
Cl |
C O |
AlCl4 |
O |
AlCl3 |
|
|
|
|
|
Сульфирование. Сульфирование ароматических соединений широко используется в промышленном органическом синтезе в производстве красителей, поверхностно-активных веществ, лекарственных препаратов. Реакция сульфирования – это введение группы SO3H в ароматические соединения. Образующиеся продукты называют сульфокислотами. В качестве сульфирующих реагентов используют 98-100%-ную серную кислоту, а также олеум, содержащий от 20 до 60% серного ангидрида, растворённого в безводной серной кислоте. Реакционная способность серной кислоты резко повышается с увеличением содержания SO3. Так, бензол, толуол, хлорбензол и бромбензол при 40 ºС реагируют с SO3 примерно в 106 раз быстрее, чем с 98%-ной серной кислотой. Олеум применяют для сульфирования инертных аренов. Обычно принимаемый механизм сульфирования включает три стадии. На стадии (1) происходит самопротонирование серной кислоты с образованием молекулы серного ангидрида (SO3), которая представляет собой электрофильную частицу. Серный ангидрид имеет дефицит электронов и, следовательно, является кислотой Льюиса. На стадии (2) электрофильный реагент присоединяется к молекуле бензола, давая промежуточный σ-комплекс. На стадии (3) происходит отрыв иона водорода с образованием аниона бензолсульфокислоты:
2H2SO4 → H3O+ + HSO4 + SO3 |
(1) |
104
SO3 + C6H6 → C6H6+ SO3 |
(2) |
C6H6+ SO3 + HSO4 → C6H5SO3 + H2SO4 |
(3) |
Так как бензолсульфокислота является сильной кислотой, то она сильно диссоциирована.
Важной особенностью реакции сульфирования является её обратимость. Ароматические сульфокислоты реагируют с перегретым водяным паром в кислой среде, образуя исходный арен и серную кислоту.
При сульфировании 98-100% серной кислотой или олеумом наблюдается кинетический изотопный эффект kH/kD в интервале от 1,15 до 1,7, т.е. определяющей скорость стадией является стадия отщепления протона от σ- комплекса. При концентрации серной кислоты ниже 95% кинетический изотопный эффект отсутствует
Реакция сульфирования подчиняется термодинамическому контролю. Так, например, при сульфировании фенола при 20 ºС образуется приблизительно равное количество орто- и параизомеров. При сульфировании фенола при 100ºС образуется 90% пара-гидроксибензолсульфокислоты:
|
OH |
|
o |
|
|
|
|
OH |
SO3H |
|
|
OH |
|||||||||||||||
|
|
|
|
H SO , |
100 |
C |
|
|
|
|
|
|
|
|
|||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||||||
|
|
2 4 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
H2O |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
10% |
|
90 % |
|
|
|
|
|
|
||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
SO3H |
|||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||||||||||
OH |
|
|
|
|
|
|
OH |
|
|
|
|
|
|
|
OH |
||||||||||||
|
|
|
|
H2SO4 , 20 oC |
|
|
|
|
|
SO3H |
|
|
|
||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||
|
|
|
|
|
|
|
|
H2O |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
49% |
|
51% |
|
|
|
|
|
|
|||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
SO3H |
|||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||||
Бензол образует бензолсульфокислоту с выходом 90% при действии 94%-ной серной кислоты при температуре 40 ºС:
Введение второй сульфогруппы требует более жестких условий. Обычно эту реакцию проводят, применяя 20% олеум при температуре 100 ºС, в этих условиях получают мета-бензолдисульфокислоту с выходом 90%.
1
2
3
4
5
6
7
8
9
105
|
|
|
|
|
|
|
D2SO4 |
|
|
|
|
|
|
|
R |
|
|
|
|
|
|
|
|
|
|
|
|
|
D |
|
|||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||||||
|
|
|
|
|
|
|
HDSO4 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
NO2 |
|
||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||
|
|
|
|
|
|
|
HNO (H SO ) |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||
3 |
2 |
4 |
|
|
|
|
|
R |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||
|
|
|
|
|
|
|
H2O |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
SO3H |
|||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||||||
|
|
|
|
|
|
|
H2SO4 |
|
|
|
|
|
|
|
R |
|
|
|
|
|
|
|
|
|
|
|
|
||||||||||
|
|
|
|
|
|
|
H2O |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||
|
|
|
|
|
|
|
Hal2 |
|
|
|
|
|
|
|
R |
|
|
|
|
|
|
|
|
|
|
|
|
|
Hal |
|
|||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||||||
|
|
|
|
|
|
|
HHal |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||
|
|
|
|
|
|
|
RCl |
|
|
|
|
|
|
|
R |
|
|
|
|
|
|
|
|
|
|
|
|
R |
|
||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||
|
|
|
|
|
|
|
HCl |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
CH2Cl |
||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||
|
|
|
|
|
|
|
H2C=O (HCl) |
|
|
R |
|
|
|
|
|
|
|
|
|
|
|
||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||
|
|
|
|
|
|
|
H2O |
O |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||
|
|
|
|
|
|
|
R |
|
C |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
C |
|
|
|
O |
|||
|
|
|
|
|
|
|
|
Cl |
|
|
|
|
|
|
R |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||
|
|
|
|
|
|
|
HCl |
|
|
|
|
|
|
|
|
|
|
|
R |
|
|||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||
|
|
|
|
|
|
|
1(Al2O3, HCl) 2 H2O |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
C |
|
|
|
|
O |
||||||||
|
|
|
|
|
|
|
HCN |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||
|
|
|
|
|
|
|
|
|
R |
|
|
|
|
|
H |
|
|||||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||||||||||||
|
|
|
|
|
|
|
NH4Cl |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
C |
|
|
|
|
O |
|||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||
|
|
|
|
|
|
|
CO (AlCl3, CuCl, HCl |
|
|
R |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
H |
|
|||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||
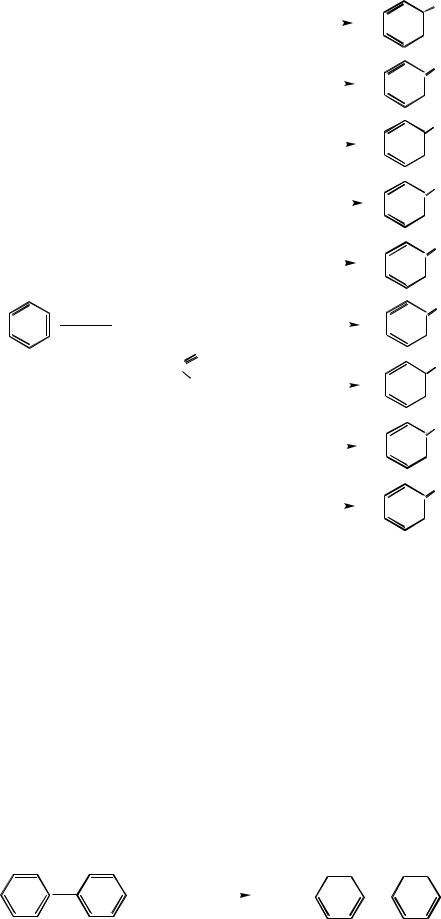
Рис.9.3. Некоторые реакции электрофильного замещения
вароматических соединениях
9.6.Электрофильное замещение в полициклических ароматических системах
Наиболее важным и интересным представителем класса полиядерных аренов является дифенил. Экспериментальные данные показывают, что дифенил в реакциях электрофильного замещения более реакционноспособен, чем бензол. Электрофильные реагенты атакуют орто- и пара-положения фенильных колец, причём преимущественно пара-положение. Наиболее вероятно, что второе бензольное кольцо проявляет положительный индуктивный, а не мезомерный эффект:
94% |
H2SO4 |
, BF3 |
HO3S |
|
|
|
|
|
|
|
|
|
|
|
SO3H |
|
|
|
|
|
|
|
|
|
|
|
|
||||||
|
|
|
|
|
|
|
|
|
|
|
||||||
|
|
H2O |
|
|
|
|
|
|
|
|
|
|||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|

106
При переходе от дифенила к флуорену, в котором оба бензольных кольца строго копланарны и их взаимное влияние более ярко выражено, скорость реакции электрофильного замещения резко возрастает. Замещение, как правило, протекает во второе положение:
Br2 |
Br |
Br |
|
HBr |
|||
|
|
||
CH2 |
|
CH2 |
Электрофильное замещение в конденсированных системах (нафталин, антрацен) может протекать как по классическому механизму SE(Ar) с образованием σ-комплекса, так и по механизму присоединения –отщепления. Доказано, что галогенирование и нитрование антрацена в мягких условиях протекает через промежуточное образование продуктов 9,10-присоединения, которые легко превращаются в 9-производные антрацена:
H |
Br |
Br |
Br2 |
|
t |
|
|
- HBr |
H |
Br |
|
При избытке электрофильного реагента образуются 9,10-дизамещённые производные антрацена.
9.7. Электрофильное замещение в небензоидных ароматических соединениях
Пятичленные ароматические гетероциклические соединения, такие, как фуран, тиофен, пиррол вступают в реакции электрофильного замещения, так же как и другие ароматические соединения.
Отличие состоит лишь в том, что они гораздо реакционноспособнее, чем бензол. По своей реакционной способности они сравнимы с фенолом и анилином. Электрофильное замещение в пирроле , фуране и тиофене происходит в положение 2, если оно занято, то замещение осуществляется в положение 3. Для реакций электрофильного замещения в ряду пятичленных гетероциклических соединений часто не требуется катализ. Так, при бромировании тиофена не требуется кислота Льюиса, а бром вступает сразу в два положения, рис.8.4, уравнение (2). Нитрование пиррола, фурана и тиофена пятичленных гетероциклов проводят с помощью ацетилнитрата, рис. 9.4 уравне-
ние (3).
На рис. 9.4 приведена схема ряда реакций электрофильного замещения тиофена:
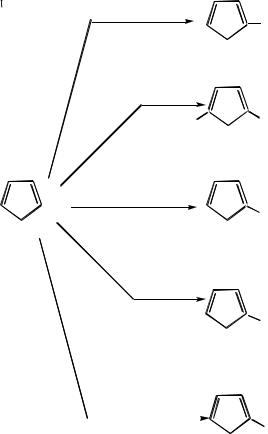
107
H2SO4 |
|
SO3H |
(1) |
|
S |
||
|
|
|
|
Br2 |
|
|
(2) |
|
|
|
|
Br |
S |
Br |
|
|
|
|
|
CH3CO2No2 |
|
(3) |
S |
|
S |
NO2 |
|
|
I2
(4)
S
CH2O, HCl |
|
|
(5) |
|
S |
CH2Cl |
|
|
|
Рис.9.4. Некоторые реакции электрофильного замещения тиофена: (1) – сульфирование; (2)
– бромирование; (3) нитрование ацетилнитратом; (4) − иодирование в присутствии оксида ртути; (5) − хлорметилирование.
Пятичленные ароматические гетероциклические соединения, такие, как фуран, тиофен, пиррол вступают в реакции электрофильного замещения, так же как и другие ароматические соединения.
Отличие состоит лишь в том, что они гораздо реакционноспособнее, чем бензол. По своей реакционной способности они сравнимы с фенолом и анилином. Электрофильное замещение в пирроле , фуране и тиофене происходит в положение 2, если оно занято, то замещение осуществляется в положение 3. Для реакций электрофильного замещения в ряду пятичленных гетероциклических соединений часто не требуется катализ. Так, при бромировании тиофена не требуется кислота Льюиса, а бром вступает сразу в два положения, рис.8.4, уравнение (2). Нитрование пиррола, фурана и тиофена пятичленных гетероциклов проводят с помощью ацетилнитрата, рис. 9.4 уравне-
ние (3).
Обобщая все экспериментальные данные по электрофильному замещению в ароматических системах можно сделать следующий вывод: реакцион-
ная способность и ориентация в реакциях электрофильного замещения в ароматических системах определяется скоростями образования возникающих в реакции карбокатионов. В свою очередь эти скорости зависят от
108
степени рассредоточения заряда вследствие действия электронодонорных или электроноакцепторных групп.
10. РЕАКЦИИ ЭЛЕКТРОФИЛЬНОГО ПРИСОЕДИНЕНИЯ К ДВОЙНЫМ УГЛЕРОД–УГЛЕРОДНЫМ
СВЯЗЯМ
Реакции алкенов и диенов обусловлены характером двойной связи, которая обладает электронодонорными свойствами. Во всех реакциях присоединения к алкенам двойная связь разрывается и две группы присоединяются, образуя две новые σ-связи, суммарная энергия которых намного превышает энергию π-связи. Многие реакции присоединения по двойной связи протекают под действием полярных или легко поляризуемых реагентов. Все эти реакции объединяет механизм их протекания. Исследованию этой группы реакций было посвящено значительное число научных работ, результатом которых стал детально разработанный механизм реакций электрофильного присоединения.
10.1.Реакции электрофильного присоединения к алкенам
Вобщем виде реакции присоединения можно представить следующеё
схемой:
H |
C |
|
C |
H |
|
|
X - Y |
|
H |
C |
|
C |
H |
||
|
|
|
|||||||||||||
H |
|
|
|
H |
|
|
|
|
H |
|
|
|
|
|
H |
|
|
|
|
|
|
|
|
|
|
X Y |
|
||||
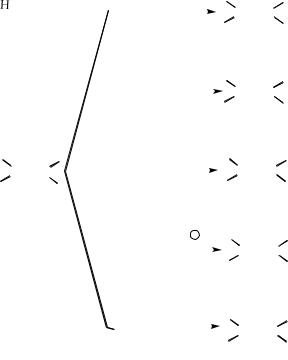
Присоединение к алкенам протекает в две стадии. На первой стадии катион или положительно заряженный конец поляризованной молекулы атакует π-электроны двойной связи, образуя карбокатион, на второй стадии карбокатион взаимодействует с нуклеофилом:
C |
C |
E Nu |
C |
C |
Nu |
(1) |
|
|
|
E |
|
|
|
C |
C |
Nu |
C |
C |
|
(2) |
E |
|
|
E |
Nu |
|
|
Скорость первой стадии в значительной степени или полностью определяет общую скорость присоединения. Первая стадия заключается в атаке электрофильным реагентом, и поэтому реакцию называют электрофильным присоединением. Электрофилом может быть любая электронодефицитная молекула.
109
Вторая стадия протекает быстро. В этой реакции электронодефицитный углерод получает пару электронов, присоединяя нуклеофил.
На схеме представлены некоторые реакций алкенов, которые подчиняются механизму электрофильного присоединения:
|
|
|
|
|
|
|
|
|
Br2 |
|
|
|
C |
|
|
|
|
C |
|||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Br |
|
|
|
|
|
|
|
|||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Br |
|||||||||||||||||
|
|
|
|
|
|
|
|
|
H2SO4 |
|
|
|
C |
|
|
|
|
C |
|||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
H |
|
SO3H |
||||||||||||||
C |
|
C |
|
|
|
|
|
|
HCl |
|
|
|
C |
|
|
|
|
C |
|||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
H |
|
|
|
Cl |
||||||||||
|
|
|
|
|
|
|
|
|
H2O(H) |
|
|
|
|
|
|
C |
|
|
|
|
C |
||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||
|
|
|
|
|
|
|
|
|
BH3 |
|
|
|
|
|
H |
|
|
|
|
|
OH |
||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
C |
|
|
|
|
C |
||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
BH2 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||
10.2. Доказательство механизма
А. Первая стадия во многих реакциях электрофильного присоединения
– это присоединение протона к алкену (гидратация в присутствии минеральных кислот, присоединение галогеноводородов). В случае атаки кислотой скорость реакции зависит от силы применяемой кислоты. Наблюдается следующая последовательность в лёгкости протекания реакции: HClO4 > H2SO4 > HX >> CH3COOH. Лёгкость присоединения галогеноводородов также связана с их кислотными свойствами.
В. При проведении реакции бромирования в растворе, содержащем помимо Br другой нуклеофил, например, Cl в реакционной смеси обнаруживается бромхлорпроизводное наряду с дибромидом, но дихлорид не образуется: