

3 курс / Фармакология / Основы_фармацевтической_технологии_Спичак_И_В_,_Автина_Н_В_2010
.pdfОСНОВЫ ФАРМАЦЕВТИЧЕСКОЙ ТЕХНОЛОГИИ
по существу означает механический акт включения уже одобренных лекарственных средств в специальный перечень (регистр или реестр). В России, в отличиеотмировойпрактики, регистрацияраспространяетсяинафармацевтические субстанции.
Регистрация фармацевтических продуктов является центральным звеном государственного регулирования лекарственного рынка по всем параметрам: по номенклатуре, эффективности, безопасности, качеству, информации для врачей и потребителей и т. д. Вместе с лицензированием производителей, импортеров и дистрибьюторов порядок регистрации служит основой системы обеспечения качества продукции в фармацевтическом секторе экономики.
К заявке на регистрацию фармацевтического продукта прилагается пакет документов (часто именуемый «регистрационное досье»), содержащий результаты исследований и испытаний препаратов и описание условий их производства. Содержание и объем регистрационного досье, а также методики проведенияисследованийпоегоподготовкеустанавливаютсянациональным органом нормативного контроля лекарств. К документам, как правило, прилагаются образцы готовой продукции в товарной (рыночной) упаковке и с окончательной маркировкой; по решению регистрационного органа они могут быть подвергнуты лабораторному анализу на соответствие предлагаемой спонсором спецификации.
Для удобства подготовки и рассмотрения регистрационное досье обычно делится на 4 основных раздела, условно именуемых: «Резюме досье» (общая информация о продукте и его производителе); «Качество» (результаты химических, фармацевтических и биологических испытаний, данные о производстве и контроле); «Безопасность» (в основном результаты доклинических исследований: изучение медико-биологических свойств на лабораторных животных, культурах тканей и т.п.) и «Эффективность» (результаты клинических испытаний).
Вслучае положительной оценки материалов, содержащихся в регистрационном досье, продукт разрешается к сбыту; заявителю (спонсору) выдается регистрационное удостоверение, обычно действительное в течение 5 лет. Удостоверение может быть отозвано или его действие может быть приостановлено в случае изменения условий, на которых оно было выдано, например при изменении условий производства данного препарата, резкого ухудшения его качества, выявления неблагоприятных побочных действий и т. п. По истечении срока действия регистрации спонсор может подать документы на продление этого срока на следующие 5 лет (перерегистрация).
Вслучае разрешения препарата к продаже важнейшие сведения из раздела «Качество» (условия производства, спецификация готового продукта, срок хранения и др.) и из раздела общей информации (порядок применения,
31

Спичак И.В., Автина Н.В.
показания, противопоказания и т.п.) автоматически утверждаются. В связи с этим производитель не имеет права отступать от изложенных в досье положений без согласия регистрационного органа. Исключение составляют «незначительные изменения».
Исследования, результаты которых представлены в разделах «Безопасность» и «Эффективность», должны быть проведены с соблюдением правил GLР и GСР, что включает возможность инспекционных и документальных проверок представленных данных. Достоверность материалов раздела «Качество», прежде всего в части условий производства, также может быть проверена в ходе предрегистрационного обследования. Такие обследования все чащепрактикуютсяорганаминормативногоконтролялекарствиндустриальных стран.
В настоящее время разделы «Безопасность» и «Эффективность», как правило, представляются лишь при регистрации препаратов, содержащих новые химические вещества. Согласно широко распространенной практике, воспроизведенные препараты регистрируются на основании так называемой «сокращенной заявки», включающей документацию по качеству, во многих случаях в сочетании с данными по биоэквивалентности. Раздел «Качество» обязателендлявсехпрепаратов, авотношениидженериковонявляетсяединственным. После регистрации нового препарата материалы разделов «Безопасность» и «Эффективность» носят в основном архивный характер; раздел «Качество» остается основой поддержания на заявленном уровне условий производства и контроля в течение всего «жизненного цикла» препарата.
Важнейшим элементом в системе регистрации являются регистрационные требования и методические указания по проведению предрегистрационных исследований и испытаний. Их содержание (в сочетании с ресурсами для приобретения медикаментов) определяет лицо лекарственного рынка той или иной страны.
Требования к заявке на регистрацию воспроизведенных препаратов (содержание и формат заявки и сопровождающих ее материалов о результатах изучения и испытаний препарата и условиях его производства) в рекомендациях ВОЗ, в документах ЕС и России отличаются. Анализ материалов и документов, представляемых вместе с заявкой на регистрацию лекарственных средств, позволяетсделатьследующиевыводы: во-первых, регистрационные требования ВОЗ и ЕС во многом совпадают. Во-вторых, соответствующие требования Российской Федерации отстают от рекомендаций ВОЗ и нормативных положений ЕС.
Регистрационные требования ВОЗ и ЕС можно суммировать следующим образом.
Технологический блок:
32

ОСНОВЫ ФАРМАЦЕВТИЧЕСКОЙ ТЕХНОЛОГИИ
•Описание процессов изготовления фармацевтической субстанции.
•Описание процессов изготовления готовой лекарственной формы.
•Валидация технологических процессов.
Контрольно-аналитический блок:
•Спецификации качества исходных продуктов и полупродуктов в изготовлении фармацевтической субстанции.
•Спецификация качества фармацевтической субстанции.
•Спецификации качества вспомогательных веществ.
•Контроль производства готовой лекарственной формы.
•Спецификация качества готовой лекарственной формы.
•Валидация аналитических методик (для рутинного анализа и для изучения стабильности).
•Результаты анализа производственных серий.
Фармацевтический блок:
•Пояснительная записка к прописи.
•Спецификации качества упаковочных материалов.
•Результаты изучения стабильности.
Эффективность, безопасность:
•Биоэквивалентность (ЕС).
•Взаимозаменяемость (ВОЗ).
Вроссийских требованиях практически полностью отсутствует «технологический блок», отсутствуют также многие элементы других разделов.
К заявке на регистрацию продуктов, содержащих новые химические соединения, как отмечалось выше, прилагаются, помимо раздела «Качество», также разделы «Безопасность» и «Эффективность»; кроме того, в разделе «Качество» требуется представление ряда дополнительных данных в отношении химических и физических свойств лекарственной субстанции, стабильности, биодоступности, прописи и свойств дозированной формы, использованной в клинических испытаниях, и т. п.
Национальные регистрационные системы и требования в различных странах имеют определенные отличия, однако существует четко выраженная тенденция их сближения (гармонизации). Практически полностью гармонизированы требования в странах ЕС, с 1990 г. ведется работа по согласованию требований между Евросоюзом, США и Японией в рамках Международной конференции по гармонизации технических требований к регистрации лекарств. Значительная работа (к сожалению, недостаточно последовательно осуществляемая) по сближению регистрационных требований на глобальном уровне проводится в рамках ВОЗ.
Отечественныерегистрационныетребованияиметодическиеуказанияпо подготовке регистрационных материалов за последние годы приблизились к
33

Спичак И.В., Автина Н.В.
общепринятым в мировой практике, однако, как показано на примере раздела «Качество» для дженериков, еще не достигли этого уровня.
2. Лицензирование предприятий по производству лекарственных средств
Лицензирование производителей фармацевтических препаратов является столь же важным элементом системы обеспечения их качества, как и регистрация продуктов. По мнению многих специалистов, лицензирование даже более важно, поскольку наличие лицензии на производство у заявителя является необходимым условием получения им регистрационных удостоверений на отдельные препараты. Этот взгляд нашел отражение в тексте руководства по GМР ВОЗ: «Зарегистрированные фармацевтические продукты должны изготовляться только лицензированными производителями» (начало раздела «Общие соображения»).
Производственная лицензия выдается на основе заявления, к которому прилагается описание производственных помещений, оборудования, сведения об организации производства, о руководящих, технических и вспомогательных работниках, а также перечень препаратов, намеченных к производству. Подача заявки сопровождается уплатой денежного сбора. Во многих странах в текст заявки включается обязательство производителя беспрепятственно допускать на предприятие государственных инспекторов для обследований. Важнейшим условием лицензирования фармацевтических производителей является соблюдение ими требований GМР, что устанавливается либо только на основе изучения представленной документации с описанием условий производства, либо, чаще всего, с учетом результатов инспекционного обследования.
В лицензии обычно указываются категории препаратов, разрешенных к производству на данном предприятии, или прилагается их перечень. Указываются также фамилии руководителей, ответственных за производство, за контроль качества продукции и за другие ключевые функции (упаковка, маркировка и т. п.). Срок действия лицензии – обычно пять лет. Лицензия может быть отозвана или ее действие может быть приостановлено в случае изменения условий, на которых она была выдана, например, при изменении номенклатуры производимых лекарств, замены ключевых руководителей, резкого ухудшения качества продукции.
Для того, чтобы избежать отзыва или приостановки действия лицензии, производитель обязан информировать лицензионный орган (обычно в составе национального органа нормативного контроля лекарств) о намечаемых или уже осуществленных изменениях. Особое место среди преднамеренных изменений условий и организации производства занимает замена специали-
34

ОСНОВЫ ФАРМАЦЕВТИЧЕСКОЙ ТЕХНОЛОГИИ
ста, ответственного за качество. Такая замена может быть осуществлена только по согласованию с лицензионным органом. Это требование обеспечивает руководителю службы качества относительную независимость от вышестоящего руководства предприятия.
В России юридической основой лицензирования является Федеральный закон«Олекарственныхсредствах» (ст. 15) иподзаконныеакты, вчастности, «Положение о порядке получения разрешения (лицензии) на промышленное производство и реализацию производителями лекарственных средств» от 3 марта 1994 г., которые в настоящее время пересматриваются.
3. Основные положения биофармации
Новым в технологии является биофармацевтическое направление, которое сложилось в самостоятельное учение в начале 60-х годов ХХ в.
Биофармация – наука, изучающая биологическое действие лекарственных препаратов в зависимости от их физико-химических свойств, лекарственной формы и технологии приготовления.
Биофармация появилась после установления фактов терапевтической неэквивалентности лекарственных препаратов, т.е. ЛП одного состава, но изготовленные разными предприятиями, отличались по эффективности. Для специалистов, занимающихся производством и анализом лекарственных препаратов, это явление было неожиданным. Все препараты соответствовали требованием фармакопеи и потому, согласно общепринятой концепции, должны были быть полностью равноценными. Объяснение этому феномену терапевтической неадекватности дала новая отрасль фармации, медицины, биологии – биофармация, знаменующая рождение биологического этапа фармации.
Особое влияние на терапевтическую эффективность лекарственных препаратов оказывают следующие факторы, которые названы фармацевтическими.
Фармацевтическими факторами являются:
–химическая модификация препарата (соль, кислота, наличие эфирных связей, комплексные соединения);
–физико-химическое состояние лекарственного вещества (форма кристалла, размеры частиц, наличие или отсутствие заряда на их поверхности
ит. д.);
–вспомогательные вещества, их природа, количество;
–вид лекарственной формы и пути введения;
–фармацевтическая технология.
Химическая модификация лекарственного вещества обязательно учитывается при разработке новых лекарственных препаратов. Она обуславливает кинетику высвобождения и всасывания лекарственного препарата.
35

Спичак И.В., Автина Н.В.
Физико-химическое состояние лекарственного вещества оказывает значительное влияние на его биологическую активность. Известна способность химических соединений иметь различную структуру, характеризующуюся в каждом конкретном случае специфической совокупностью свойств. Геометрическаяформаисоставобразующихсякристалловсущественнозависятот характера растворителя, скорости кристаллизации, температуры процесса, от примесей, величины давления и др. факторов. Считают, например, что 3060% сульфаниламидов, 70% барбитуратов полиморфны, 1/3 всех органических соединений имеет по крайней мере две кристаллические формы. Накопленодостаточноеколичествоэкспериментальногоматериалаозависимости структуры веществ и их биологической доступности. Вопросам измельчения в фармацевтической технологии придается особое значение. Известно, что с уменьшением размера частиц резко увеличивается поверхностная энергия измельчаемого лекарственного вещества. При тонком измельчении лекарственные вещества лучше растворяются, быстрее и полнее участвуют в химическихреакцияхит. д. Измельчениеможетсущественнымобразомвлиять на терапевтическую активность лекарственных веществ вследствие измененияпроцессовихвсасывания. Этопроисходитприизменениирастворимости лекарственных веществ, скорость которой прямо пропорциональна площади поверхности и обратно пропорциональна величине частиц вещества. Например, при назначении одинаковых доз сульфадимезина микронизированного
иполученного в заводском производстве без дополнительного измельчения выявлено, что в первом случае в плазме крови людей содержание вещества на 40% выше, максимальная концентрация достигается на 2 часа раньше, а общее количество всосавшегося вещества на 20% больше, чем во втором случае. При уменьшении размера частиц кислоты ацетилсалициловой до микронизированных увеличилось приблизительно в 2 раза ее анальгетическое, жаропонижающее и противовоспалительное действие. В аптечной практике необходимый размер частиц порошка получают при соблюдении следующих условий измельчения: выбор ступки, время измельчения, применение аппаратов, порядок смешивания, особые правила и приемы технологии.
Не менее важное значение в технологии лекарственных форм имеет правильный выбор вспомогательных веществ. До самого недавнего времени во вспомогательных веществах видели только индифферентные формообразователи, значение которых сводились к приданию соответствующей формы
иобъема лекарственному веществу с целью удобства его приема, транспортировки, хранения. Однако открытия последних десятилетий привели к осознанию биологической роли вспомогательных веществ. Они могут усиливать, снижать действие лекарственных веществ или изменять его характер под влиянием различных причин (комплексообразование, молекулярные реакции и др.).
36

ОСНОВЫ ФАРМАЦЕВТИЧЕСКОЙ ТЕХНОЛОГИИ
Применение тонких высокочувствительных методов анализа препаратов (газожидкостная, тонкослойная хроматография, рентгеноструктурный анализ, спектрофотометрия) позволили установить самые тесные взаимоотношения лекарственных и вспомогательных веществ. Такие обычно применяющиеся вспомогательные вещества, как желатин, крахмалы, полиэтиленоксиды, производные целлюлозы, неионоактивные ПАВ, способны вступать в реакции взаимодействия (в частности, комплексообразование) с лекарственными веществами самой различной природы, образуя соединения, характеризующиеся иными, чем исходные вещества, свойствами. В качестве примера рассмотрим влияние вспомогательных веществ на активность лекарственных веществ в мазях и суппозиториях.
Среди факторов, влияющих на высвобождение лекарственных веществ в мазях, наибольшеевниманиеуделяютоснове. Влияниетипаосновыразлично в зависимости от способа введения лекарственного вещества. Установлено, например, что кислота борная не оказывает бактериостатического действия при использовании жировых основ, но эффективна при изготовлении мазей на гидрофильных основах, в которых содержится большое количество воды. По-видимому, терапевтическое действие проявляет образующийся раствор кислоты борной. Йод, напротив, малоактивен в основах, содержащих большое количество воды.
Введение в состав мазевых и суппозиторных основ эмульгаторов, ПАВ и др. активаторов всасывания является одним из важных факторов, оказывающих влияние на активность лекарственных веществ. Натрия лаурилсульфат способствует увеличению резорбции микрокристаллического сульфапиридазина из гидрофильной основы. Показана способность диметилсульфоксида легко проникать через неповрежденную кожу, транспортировать, депонировать и пролонгировать при этом поступление лекарственных веществ в организм.
Перспективным вспомогательным веществом в технологии мазей, суппозиториев, растворов для инъекций, глазных лекарственных пленок и др. лекарственных форм является коллаген. Предполагается, что лекарственное вещество, попадая в «петли» молекул коллагена, образует соединение, обеспечивая тем самым пролонгированное действие. Вспомогательные вещества должны отвечать основному требованию – раскрывать всю гамму фармакологических свойств препарата, обеспечивать оптимальное действие лекарственного вещества. Правильный выбор вспомогательных веществ позволяет снизить концентрацию лекарственного вещества при сохранении терапевтического эффекта.
Значение лекарственной формы. Оптимальная активность лекарственного вещества достигается только назначением его в рациональной, научно
37

Спичак И.В., Автина Н.В.
обоснованнойлекарственнойформе. Выборлекарственнойформыопределяет и способ введения лекарственного вещества в организм. Эффективность лекарственного вещества зависит от того, какой путь совершит оно до того, как попадет в кровь. При ректальном способе лекарственное вещество попадает в кровь, минуя печень, и не подвергается химическому воздействию ее ферментов, желудочногосокаижелчи. Поэтомуприректальномприменении лекарственное вещество всасывается через 7 мин., а при пероральном – через 30 мин.
Значение технологических факторов. Способ получения ЛФ во многом определяет стабильность препарата, скорость его высвобождения из ЛФ, интенсивность всасывания – и в целом терапевтическую эффективность. Например, выбор способа гранулирования таблеток обуславливается сохранностью лекарственного вещества в ЛФ. Перспективны технологии многослойных таблеток и спансул (желатиновых капсул, наполненных гранулами) для обеспечения пролонгированного и дифференцированного действия.
4. Биологическая доступность
Выявление терапевтической неадекватности препаратов обусловило изменение взглядов как на процесс изготовления, так и на оценку качества лекарственных препаратов. Стало очевидным, что нельзя только на основании показателей, принятых нормативно-технической документацией, включая количественное содержание действующего вещества, получить полную информацию о возможном изменении его активности в лекарственной форме. Для этого необходимо знать, как ведет себя лекарственное вещество в условиях организма, т. е. знать его биологическую доступность.
Биологическая доступность (БД) определяется долей всосавшегося в кровь лекарственного вещества от общего содержания его в соответствующей лекарственной форме, продолжительностью нахождения его определенной концентрации в организме.
Степень БД определяют в сравнении со стандартной лекарственной формой, которая хорошо всасывается. При этом используют одинаковые дозы стандартной и исследуемой лекарственной формы. БД выражается в процентах и может быть представлена в виде следующего уравнения:
БД = В / А · 100, где А и В – количество всосавшегося лекарственного вещества из стандартной и исследуемой лекарственных форм соответственно.
Различают абсолютную и относительную БД. В качестве стандартной лекарственной формы при определении абсолютной БД применяют раствор для внутривенного введения.
На практике чаще определяют относительную БД, когда стандартом являетсяхорошовсасывающаясяпероральнаялекарственнаяформа(например,
38

ОСНОВЫ ФАРМАЦЕВТИЧЕСКОЙ ТЕХНОЛОГИИ
раствор). При фармакокинетическом методе определения БД производят последовательный забор проб биожидкостей в течение строго определенного времени, а затем с помощью наиболее точных и чувствительных аналитических методов определяют в пробах концентрацию лекарственного вещества. На основании полученных данных (содержание веществ или их метаболитов) строят графики, отражающие кинетику того или иного лекарственного вещества во времени, и с помощью фармакокинетических методов рассчитывают БД.
Исследованияin vivo невозможныдлямассовойоценкикачествапрепаратоввпроизводственныхусловиях. Впрактикеимеетместовесьмачастая, хотя и необязательная, корреляция между степенью БД и скоростью растворения лекарственных веществ. Это позволяет в первом приближении использовать «тест растворения» для характеристики БД препарата. Доступность, определяемую in vitro, часто называют, в отличие от биологической, фармацевтической. Существует несколько методов определения скорости растворения, которыеклассифицируютисходяизобъемасреды, ееподвижности, значения рН и других физических показателей. Многими фармакопеями мира, в том числе и российской, для определения скорости растворения принят прибор типа «вращающаяся корзинка». В настоящее время внедряют приборы, позволяющие проводить исследования препаратов в условиях, близких к условиям желудочно-кишечного тракта, и характеризовать не только процессы высвобождения лекарственных веществ, но и процессы всасывания. Такие приборы и тесты открывают широкие возможности для установления БД во время разработки новых лекарственных форм и проведения текущей проверки их качества в контрольно-аналитических лабораториях.
1.4. Измельчение, просеивание, перемешивание твердых материалов
1.Измельчение. Характеристика процесса.
2.Машины по измельчению минерального сырья. Устройство и принцип работы.
3.Аппаратура для измельчения лекарственного растительного сырья
4.Просеивание. Характеристика процесса. Механизированные сита. Устройство и принцип работы.
5.Смешивание. Характеристика процесса. Аппаратура для смешивания лекарственных и вспомогательных веществ.
1. Измельчение. Характеристика процесса
Процессы измельчения, просеивания, смешивания широко применяются дляпроизводствалекарственныхформ: порошков, сборов, таблеток, настоек, экстрактов, мазей и др.
39

Спичак И.В., Автина Н.В.
Измельчение - процесс уменьшения размера кусков или частиц твердых материалов путем механического воздействия.
Физические основы измельчения. Процесс измельчения связан со значительной затратой энергии. При измельчении материала происходит деформация.
Виды деформации:
–упругие деформации (не происходит изменение формы тела);
–пластические деформации (приводит к разрушению тела).
Кратко охарактеризуем теории измельчения, поясняющие расход энергии в процессе измельчения твердых тел.
Теория Риттингера (поверхностная теория). Согласно этой теории работа по измельчению затрачивается на образование новых поверхностей материала: А=К•  F,
F,
 F – прирост новой поверхности; К – коэффициент пропорциональности, который определяют опытным путем. Коэффициент обозначает расход энергии на единицу вновь образуемой поверхности.
F – прирост новой поверхности; К – коэффициент пропорциональности, который определяют опытным путем. Коэффициент обозначает расход энергии на единицу вновь образуемой поверхности.
Теория Кирпичева-Кика (объемная теория) – работа по измельчению расходуется на деформацию кускового материала до момента его разрушения и затрачивается на образование новых объемов материала: А = k • 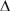 V
V
 V- прирост новых объемов;
V- прирост новых объемов;
k – коэффициент пропорциональности, который обозначает энергию или работу, затрачиваемую на преодоление сил упругой и пластичной деформации при образовании новой единицы объема материала.
Теория Ребиндера (объединенная теория) – работа по измельчению, затрачивается на образование новых поверхностей и на образование новых объемов.
A = K •  F + k •
F + k • 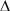 V
V
Увеличение поверхности твердых тел, участвующих в технологическом процессе, ускоряет эти процессы, увеличивает выход и улучшает качество продукта.
Способы измельчения твердых тел:
1)раздавливание;
2)раскалывание;
3)размалывание;
4)резание;
5)распиливание;
6)истирание;
7)жесткий удар;
8)свободный удар.
40