

2 курс / Гистология / Врожденные_пороки_сердца_Зиньковский_М_Ф_,_Возианов_А_Ф_ред_
.pdf102 |
Часть 1. ОБЩИЕ СВЕДЕНИЯ О ВРОЖДЕННЫХ ПОРОКАХ СЕРДЦА |
|
|
|
|
I
II
III
IV
Рис. 2. Пример родословной с аутосомно доминантным типом наследования АВСД (явление неполной пенетрантности)
Первой задачей при анализе родословной явля ется установление наследственного характера признака. О наследственной природе признака (заболевания) можно думать, если признак встре чается в родословной несколько раз и исключается воздействие сходных внешних факторов во время всех беременностей.
Следует помнить, что наличие двух или более случаев ВПС в родословной не является прямым доказательством исключительно наследственной обусловленности порока (подобная картина наб людается и при мультифакториальном наследова нии), а присутствие порока сердца только у про банда не исключает его наследственной природы.
После составления графической схемы родос ловной устанавливается тип наследования. Как правило, это делает врач генетик в медико генети ческой консультации или центре с последующим расчетом генетического риска в каждом отдельном случае. Ориентировочно можно учитывать следую щие характеристики различных типов наследова ния (табл. 1).
Наследственно обусловленные ВПС
Наследственно обусловленные ВПС представ ляют собой гетерогенную группу и включают:
1. Моногенные ВПС (причиной возникновения служит мутация одного гена), которые наследу ются согласно законам Менделя. Они представ лены:
•изолированными пороками сердца при отсутствии врожденных пороков или аномалий развития других органов и систем. Так, описаны случаи аутосомно доминантного наследования мута ции специфичного гена Nkx2 5 на хромосоме
5q35, которая является ответственной за разви тие вторичного ДМПП и нарушение AV прово димости;
•синдромами множественных врожденных пороков развития (МВПР), в которых ВПС является од ним из составляющих синдром признаков. Мо ногенное наследование касается всего синдро ма как единого целого, однако проявление мо жет быть у разных членов семьи неполным и ко лебаться от одного единственного признака (необязательно ВПС) до разной комбинации составляющих этот синдром врожденных поро ков развития.
2.Хромосомные синдромы, причиной возникнове ния которых служит изменение количества и/или структуры хромосом.
Если ВПС являются частью генного или хромо
сомного синдрома, у подавляющего большинства пациентов имеются и другие пороки развития, на рушения физического и психического развития различной степени тяжести, нарушения метабо лизма и/или другая сопутствующая патологию. По данным проведенных исследований, до 66% врож денных пороков развития у плодов и мертворож денных детей носит характер множественных (30).
Врожденные пороки сердца описаны как кар динальный признак многих моногенных синдро мов. Основные из них представлены в табл. 2.
Следующие признаки позволяют заподозрить наличие у пациента моногенного синдрома:
1)семейный характер заболевания (т.е. нали чие в семье сходных случаев заболевания, в том числе лишь отдельных симптомов);
2)наличие у пациента редко встречающихся специфических симптомов или их сочетания (например, необычный запах от пациента, изме
Глава 7. ГЕНЕТИЧЕСКИЕ АСПЕКТЫ ВРОЖДЕННЫХ ПОРОКОВ СЕРДЦА |
103 |
|
|
|
|
Таблица 1. Основные характеристики разных типов наследования
Аутосомно доминантный тип наследования |
Аутосомно рецессивный тип наследования |
|
|
1. Болезнь встречается в каждом поколении родословной. |
1. Родители больных детей обычно клинически здоровы. |
2. Соотношение больных и здоровых приближается к 1:1. |
2. Чем больше детей в семье, тем чаще встречается более |
3. У здоровых потомков больных родителей рождаются |
одного больного ребенка. |
здоровыми все дети. |
3. Чем реже встречается мутантный ген в популяции, тем |
4. Соотношение больных мальчиков и девочек равное. |
чаще родители больного ребенка являются кровными |
5. У больных мужчин и женщин рождаются больные дети |
родственниками разной степени родства. |
обоих полов — как мальчики, так и девочки. |
4. Если больны оба супруга, то все дети будут больными. |
6. Чем тяжелее болезнь и чем больше она отражается на |
5. В браке больного со здоровым рождаются здоровые |
репродукции, тем больше пропорция спорадических |
дети (если здоровый не гетерозиготен). |
(единичных в родословной) случаев (новых мутаций) |
6. В браке больного с гетерозиготным носителем |
|
рождается около 50% больных детей, что имитирует |
|
доминантный тип наследования. |
|
7. Оба пола поражаются одинаково часто |
|
|
Х сцепленное доминантное наследование |
Х сцепленное рецессивное наследование |
|
|
1. Поражаются и мужчины и женщины, |
1. Болеют только мальчики. |
но больных женщин в 2 раза больше, чем мужчин. |
2. Около 2/3 случаев являются следствием передачи |
2. У больных женщин в среднем рождается 50% больных |
патологического гена от матерей носителей, |
сыновей и 50% больных дочерей. |
1/3 — являются результатом новых мутаций. |
3. У больного мужчины рождаются все больные дочери и |
3. В унаследованных случаях у больных мальчиков могут |
все здоровые сыновья. |
быть больные братья и дяди по материнской линии. |
4. В среднем женщины болеют менее тяжело, |
4. Новые мутации являются спорадическими или |
чем мужчины |
изолированными случаями. |
|
5. У здоровых мужчин не рождаются больные дети |
|
|
Y сцепленный тип наследования |
Митохондриальная наследственность |
|
|
Признак наблюдается только у лиц мужского пола и толь |
1. Болезнь передается только от матери. |
ко у потомков мужчины, который имеет этот признак |
2. У больных матерей рождаются больные как дочери, |
|
так и сыновья. |
|
3. У больных отцов рождаются здоровые как дочери, |
|
так и сыновья |
|
|
Таблица 2. Некоторые синдромы, ассоциированные с ВПС
Синдром |
Тип наследования |
Порок сердца |
Основные признаки |
|
|
|
|
Апера синдром |
Аутосомно |
СЛА, ДМЖП |
Краниосиностоз, плоское лицо, гипертелоризм, |
(рис. 3) |
доминантный |
|
синдактилия, глухота, пороки почек |
|
|
|
|
Кардио |
Спорадический |
СЛА, ДМПП |
Сходен с синдромом Нунан, редкие курчавые |
фациокутанный |
|
|
волосы, макроцефалия с битемпоральным |
синдром |
|
|
сужением, грубое лицо, ихтиоз, низкий рост, |
|
|
|
задержка развития |
|
|
|
|
CHARGE |
Спорадический |
ДМЖП, АВСД, тетрада |
Колобома глаз, порок сердца, атрезия хоан, |
ассоциация |
|
Фалло и др. |
задержка психофизического развития, гипопла |
|
|
|
зия гениталий, ушные аномалии с/либо без глу |
|
|
|
хоты |
|
|
|
|
Синдром |
Аутосомно |
ДМЖП |
Синофриз, тонкие губы, опущенные книзу углы |
Корнелии де |
доминантный, |
|
рта, грубая задержка умственного развития, |
Ланге |
спорадический |
|
низкий рост, незавершенный поворот кишечни |
|
|
|
ка, микрогнатия, редукционные пороки верхних |
|
|
|
конечностей |
|
|
|
|
104 |
Часть 1. ОБЩИЕ СВЕДЕНИЯ О ВРОЖДЕННЫХ ПОРОКАХ СЕРДЦА |
|
|
|
|
Продолжение таблицы 2
Синдром |
Тип наследования |
Порок сердца |
Основные признаки |
|
|
|
|
Анемия Фанкони |
Аутосомно |
Различные |
Низкий рост, маленькие глаза, клювовидный |
|
рецессивный |
|
нос, гипоплазия большого пальца, |
|
|
|
панцитопения, гипоплазия селезенки, |
|
|
|
аномалии почек, гиперпигментация |
|
|
|
|
Фронтоназальная |
Чаще |
Тетрада Фалло |
Гипертелоризм, широкая переносица, |
дисплазия |
спорадический |
|
срединная расщелина лица, низкий рост |
|
|
|
|
Синдром |
Спорадический, |
Тетрада Фалло |
Гемифациальная микросомия, эпибульбарный |
Гольденхара |
иногда аутосомно |
|
дермоид, преаурикулярные привески, глухота, |
(рис. 4) |
доминантный |
|
колобомы век, несращение нёба, аномалии |
|
|
|
позвонков |
|
|
|
|
Синдром Горлина |
Аутосомно |
Опухоли сердца |
Макроцефалия, выступающие лобные бугры, |
|
доминантный |
|
прогнатизм, множественные базальноклеточные |
|
|
|
невусы, карциномы, ямки на ладонях и |
|
|
|
подошвах, умственная отсталость |
|
|
|
|
Синдром |
Аутосомно |
Чаще дефекты перегоро |
Трехфаланговый или гипопластичный большой |
Холта–Орама |
доминантный |
док, в некоторых случаях |
палец кисти, гипоплазия 1 й пястной кости, |
|
|
аритмии |
лучевой кости или целой верхней конечности, |
|
|
|
узкие плечи |
|
|
|
|
Синдром |
Аутосомно |
Декстропозиция сердца, |
Цилиарная дискинезия, бронхоэктазы, глухота, |
Картагенера |
рецессивный |
обратное расположение |
situs viscerus inversus |
|
|
крупных сосудов |
|
|
|
|
|
Синдром |
Аутосомно |
Дилатация корня аорты, |
Разболтанность суставов, искривление нижних |
Ларсена |
доминантный |
аневризма, дефекты |
конечностей при рождении, плоское лицо, |
|
|
перегородок |
вдавленная переносица, несращение нёба, |
|
|
|
низкий рост |
|
|
|
|
Leopard |
Аутосомно |
Стеноз легочной артерии |
Множественные лентиго, электрокардиографи |
синдром |
доминантный |
|
ческие изменения, глазной гипертелоризм, |
|
|
|
аномалии развития гениталий, задержка |
|
|
|
развития, глухота |
|
|
|
|
Синдром |
Аутосомно |
Дилатация аорты, |
Арахнодактилия, подвывих хрусталика, |
Марфана |
доминантный |
недостаточность клапанов |
высокий рост, долихостерномелия, сколиоз, |
|
|
|
грыжи, высокое нёбо |
|
|
|
|
Noonan синдром |
Аутосомно |
Клапанный стеноз легоч |
Низкий рост, задержка развития, антимонголо |
|
доминантный |
ной артерии, ДМПП, |
идный разрез глаз, низко расположенные |
|
|
гипертрофическая |
ушные раковины, короткая шея, крипторхизм, |
|
|
кардиомиопатия |
деформации грудной клетки |
|
|
|
|
Смита–Лемли– |
Аутосомно |
ДМПП, ДМЖП, |
Умственная отсталость, микроцефалия, птоз, |
Опитца синдром |
рецессивный |
тетрада Фалло и др. |
косоглазие, открытые кпереди ноздри, синдак |
|
|
|
тилия II–III на стопах, гипоспадия, повышенный |
|
|
|
уровень сывороточного 7 дегидрохолестерола |
|
|
|
|
Тригоноцефалии С |
Гетерогенный, |
ОАП, ДМЖП, |
Умственная отсталость, выступающий мето |
(Опитца С, |
в некоторых семьях |
митральный стеноз |
пионический шов, широкие альвеолярные отро |
синдром |
аутосомно |
|
стки, монголоидный разрез глаз, телекант, ка |
|
рецессивный |
|
пиллярные гемангиомы |
|
|
|
|
VACTERL |
Спорадический |
Различные пороки, |
Дисгенезия позвонков, атрезия ануса, врожден |
ассоциация |
|
чаще ДМЖП |
ные пороки сердца, трахеопищеводный свищ, |
|
|
|
атрезия пищевода, пороки почек, дисплазия лу |
|
|
|
чевой кости |
|
|
|
|
Синдром |
Аутосомно |
Наиболее типичны ДМЖП |
Седая прядь волос, нейросенсорная глухота, |
Ваарденбурга |
доминантный с |
|
гетерохромия радужки, гипертелоризм, |
|
различной |
|
редко несращение неба, болезнь Гиршпрунга |
|
экспрессивностью |
|
|
|
|
|
|

Глава 7. ГЕНЕТИЧЕСКИЕ АСПЕКТЫ ВРОЖДЕННЫХ ПОРОКОВ СЕРДЦА |
105 |
|
|
|
|
Продолжение таблицы 2
Синдром |
Тип наследования |
Порок сердца |
Основные признаки |
|
|
|
|
Синдром |
Неизвестен |
Декстрокардия, АВСД, атре |
Правый изомеризм, аспления, |
Ивемарка |
|
зия легочной артерии, ТМА |
незавершенный поворот кишечника |
|
|
|
|
Синдром |
Аутосомно |
ДМЖП, ОАП, |
Глубокая мышечная гипотония, высокий лоб, |
Цельвегера |
рецессивный |
коарктация аорты |
плоское лицо, гепатоспленомегалия, поликистоз |
|
|
|
почек, аномалии половых органов, |
|
|
|
пороки головного мозга |
|
|
|
|
Примечание.
Более подробная информация представлена в соответствующих руководствах и электронных базах данных:
1.McKusick V.A. Mendelian Inheritance in Man. The Johns Hopkins University School of Medicine.
2.On line Mendelian Inheritance in Man (OMIM).
3.Козлова С.И. Наследственные синдромы и медико генетическое консультирование. — М., 1996.
4.London Dysmorphology Database and London Neurologenetics Database. Winter R.M., Baraitser M. Oxford University Press и др.
нение цвета мочи, голубые склеры, грубые черты лица и т.д.);
3)вовлечение в патологический процесс сразу нескольких органов и систем;
4)устойчивость к наиболее распространенным методам терапии.
Лабораторная диагностика моногенного синд рома в основе своей может быть направлена на:
•выявление этиологической причины, т.е. опре деление конкретной мутации (применяются прямые и косвенные молекулярно генетичес кие методы);
•выявление аномального первичного продукта мутантного гена (биохимические и иммуноло гические методы);
•регистрацию специфических метаболитов изме ненного обмена.
Рис. 3. Ребенок с синдромом Апера
В большинстве случаев моногенные синдромы с ВПС относятся к группе синдромов с МВПР, и для их диагностики применяется клинико генеалоги ческий метод с синдромологическим анализом (многие формы наследственной патологии прояв ляются настолько специфическим фенотипом, что клинический анализ с синдромологическим подхо дом позволяет поставить точный диагноз). Также применяются молекулярно генетические методы
— прямые, в случае известной нуклеотидной после довательности патологического гена, и непрямые, когда нуклеотидная последовательность еще не из вестна и вместе с тем имеется информация об отно сительном положении гена на генетической карте. Биохимические методы применяются редко, одна ко в некоторых случаях они являются ведущими. Так, например, биохимический метод лежит в ос нове подтверждающей диагностики синдрома Смита–Лемли–Опитца, в спектре клинического проявления которого присутствует ВПС. Для диф ференциальной диагностики применяются и цито генетические методы, так как генные синдромы с МВПР и хромосомные синдромы в ряде случаев имеют сходную клиническую картину.
Медико генетическое консультирование при моногенных синдромах основывается на установ лении типа наследования данного заболевания с последующим расчетом генетического риска для сибсов (т.е. риск повторного рождения больного ребенка у родителей пробанда) и потомков (т.е. риск иметь данное заболевание для детей пробан да) с определением наиболее эффективного спосо ба профилактики.
Подавляющее большинство ассоциаций (VAC TERL, CHARGE) являются спорадическими слу чаями, т.е. риск для сибсов не превышает общепо пуляционный.

106 |
Часть 1. ОБЩИЕ СВЕДЕНИЯ О ВРОЖДЕННЫХ ПОРОКАХ СЕРДЦА |
|
|
|
|
Рис. 4. Ребенок с синдромом Гольденхара
Хромосомные синдромы
Хромосомные синдромы — группа врожденных патологических состояний, в основе которых ле жат геномные (изменения количества хромосом) или хромосомные (изменения структуры хромо сом) мутации.
Геномные мутации, или количественные изме нения хромосомного набора, могут быть двух ти пов:
1)полиплоидии — увеличение числа хромосом, кратное гаплоидному набору хромосом (n=23);
2)анеуплоидии — увеличение или уменьшение числа хромосом в наборе, некратное гаплоидному.
К хромосомным мутациям относятся структур ные перестройки одной или двух и более хромо сом, которые могут затрагивать всю хромосому или ее часть. С точки зрения цитогенетики, струк турные перестройки хромосом классифицируются по принципу линейной последовательности рас положения генов:
•делеции (нехватка);
•инверсии (поворот на 1800);
•инсерции (вставки);
•транслокации (обмен сегментами между двумя или более хромосомами);
•кольцевые хромосомы;
•изохромосомы.
По данным Schinzel (28), ВПС относятся к од ним из частых пороков развития, сопровождаю щих хромосомные аномалии. Согласно базе дан ных CARIS (1998–2001), у 12% детей с ВПС име ются те или иные хромосомные аномалии. В 70% случаев это трисомии по хромосомам 21, 18 или 13.
Хромосомный синдром у пациента с ВПС мож но заподозрить по следующим признакам:
•врожденные пороки других органов и систем;
•задержка психомоторного и физического разви тия;
•задержка речевого развития, нарушение процес са познания, необычное поведение, нарушение внимания с гиперактивностью и другие нару шения психологического статуса;
•наличие у пациента необычных черт лица, что делает его непохожим на родителей и сибсов, особенно при наличии отставании в развитии;
•малая масса пациента при рождении, преждев ременные роды или признаки незрелости при родах в срок;
•наличие хромосомных аномалий у родителей или сибсов;
•дополнительным критерием может быть нали чие в анамнезе родителей длительного беспло дия, самопроизвольных абортов на ранних сро ках, мертворождений.
Выявление вышеописанных признаков требует направления пациента в медико генетический центр и идентификации хромосомных аномалий цитогенетическим методом. Основой данного ме тода является исследование кариотипа человека. Кариотип — это совокупность морфологических особенностей полного хромосомного набора, ти пичная для клеток представителя данного биоло гического вида. Специфичность кариотипа опре деляется общим числом хромосом, их размером и формой, а количественная и структурная стабиль ность хромосом является важнейшим условием формирования фенотипически нормального орга низма в ходе индивидуального развития. У челове ка диплоидный набор хромосом в соматических клетках составляет 46. Целью хромосомного ана лиза в клинической цитогенетике является оценка

Глава 7. ГЕНЕТИЧЕСКИЕ АСПЕКТЫ ВРОЖДЕННЫХ ПОРОКОВ СЕРДЦА |
107 |
|
|
|
|
кариотипа с выявлением возможных количествен ных или структурных аномалий хромосом путем анализа метафазных или прометафазных препара тов хромосом и правильная запись кариотипа в со ответствии с унифицированной системой записи и символизации (ІSCI, 1995).
Синдром Дауна является наиболее частым хро мосомным синдромом у пациентов с ВПС (трисо мия 21) (рис. 5). Синдром хорошо описан в много численных руководствах. Основные признаки представлены в табл. 3. Пороки сердца присут ствуют более чем у 50% детей с синдромом Дауна и являются основной причиной гибели этих паци ентов (13). Наиболее типичными являются АВСД (более половины всех случаев пороков сердца у де тей с синдромом Дауна), ДМЖП, ДМПП, ОАП, аберрантная подключичная артерия.
Синдром Патау. Причиной является трисомия по хромосоме 13 (рис. 6). Синдром сопровождает ся МВПР развития и ранней гибелью пациентов. Средняя продолжительность жизни составляет 2,5 дня, менее чем 3% пациентов доживают до 6 мес (15, 23).
Наиболее характерной комбинацией врожден ных пороков развития является сочетание постак сиальной полидактилии, несращения губы и неба, микрофтальмии, колобомы глаз и голопрозэнце фалии. Кроме того, специфическими являются врожденные пороки сердца и мочеполовой систе мы. Детальный список симптомов представлен в табл. 4.
Таблица 3. Наиболее характерные клинические признаки синдрома Дауна (Лазюк, 1979)
Порок или признак |
Частота |
|
встречаемости, % |
||
|
||
|
|
|
Брахицефалия |
81,1 |
|
|
|
|
Монголоидный разрез глаз |
79,8 |
|
|
|
|
Эпикант |
51,4 |
|
|
|
|
Плоская спинка носа |
65,9 |
|
|
|
|
Узкое нёбо |
58,8 |
|
|
|
|
Диспластичные ушные раковины |
43,2 |
|
|
|
|
Короткие и широкие кисти |
64,4 |
|
|
|
|
Клинодактилия мизинца |
56,3 |
|
|
|
|
Деформация грудной клетки |
26,9 |
|
|
|
|
Пятна Брушвильда |
68,4 |
|
|
|
|
Помутнение хрусталика |
32,2 |
|
|
|
Синдром Эдвардса. Врожденные пороки сердца являются характерным признаком и третьей, наи более частой, трисомии — трисомии по хромосоме 18 (синдром Эдвардса) (рис. 7). Как и синдром Па тау, синдром Эдвардса относится к полулетальным синдромам. Более 90% пациентов умирают, не до живая до полугода (23).
Основными признаками синдрома Эдвардса являются: резкая пренатальная гипоплазия, доли
1 |
|
2 |
3 |
|
4 |
5 |
6 |
7 |
8 |
9 |
10 |
11 |
12 |
13 |
14 |
|
15 |
16 |
17 |
18 |
19 |
20 |
|
21 |
22 |
Х |
Y |
|
|
|
а |
|
|
б |
Рис. 5. Кариотип пациента с трисомией по хромосоме 21. Стрелка указывает на дополнительную хромосому 21 (а) ; лицо ребенка с синдромом Дауна (б)

108 |
Часть 1. ОБЩИЕ СВЕДЕНИЯ О ВРОЖДЕННЫХ ПОРОКАХ СЕРДЦА |
|
|
|
|
Таблица 4. Основные врожденные пороки при синдроме Патау (Лазюк, 1979)
|
Относительная |
Пораженная система и порок |
частота |
|
пороков, % |
|
|
Лицо и мозговой череп |
96,5 |
• Низко расположенные |
80,7 |
деформированные ушные раковины |
|
• Несращения верхней губы и неба |
68,7 |
• Микрогения |
32,8 |
• Дефекты скальпа |
30,8 |
|
|
Опорно двигательный аппарат |
92,6 |
• Полидактилия кистей |
49,0 |
• Полидактилия стоп |
35,7 |
• Стопа качалка |
30,3 |
|
|
ЦНС |
83,3 |
• Аринэнцефалия |
63,4 |
• Микроцефалия |
58,7 |
• Гипоплазия мозжечка |
18,6 |
|
|
Глазное яблоко |
77,1 |
• Микрофтальмия |
70,5 |
• Колобома радужки |
35,3 |
• Анофтальмия |
7,5 |
|
|
|
Относительная |
Пораженная система и порок |
частота |
|
пороков, % |
|
|
Сердечно сосудистая система |
79,4 |
• ДМЖП |
49,3 |
• ДМПП |
37,6 |
• Пороки крупных сосудов |
42,8 |
|
|
Органы пищеварения |
50,6 |
• Незавершенный поворот кишечника |
41,6 |
• Грыжа пуповины |
19,4 |
• Гетеротопия сегментов селезенки в |
49,3 |
pancreas |
|
|
|
Мочевыводящая система |
60,6 |
• Гидронефроз |
15,1 |
• Кисты почек |
42,8 |
• Атрезия и стеноз мочеточника |
5,3 |
|
|
Половые органы |
71,6 |
• Крипторхизм |
73,2 |
• Гипоспадия |
14,8 |
• Удвоение матки и влагалища |
46,8 |
|
|
1 |
2 |
3 |
|
|
4 |
5 |
|
6 |
7 |
8 |
9 |
10 |
11 |
12 |
|
13 |
14 |
15 |
|
16 |
17 |
18 |
|
19 |
20 |
|
21 |
22 |
X |
Y |
а |
|
|
|
|
|
б
Рис. 6. Кариотип пациента с трисомией 13. Стрелка указывает на дополнительную хромосому 13 (а); новорожденный с синд ромом Патау (б) (Schinzel, 1979)

Глава 7. ГЕНЕТИЧЕСКИЕ АСПЕКТЫ ВРОЖДЕННЫХ ПОРОКОВ СЕРДЦА |
109 |
||
|
|
|
|
|
|
|
|
хоцефалия, открытый метопионический шов, на |
брыжейка, единственная подковообразная почка, |
||
висающий затылок, низко расположенные, роти |
поликистоз почек, гидронефроз. Почти у 10% па |
||
рованные кзади, диспластические ушные ракови |
циентов наблюдается аплазия лучевой кости и/или |
||
ны, маленький рот, микрогнатия, короткая груди |
большого пальца кисти (28). Основными пороками |
||
на, флексорное положение конечностей, пере |
развития, позволяющими заподозрить наличие |
||
крывание третьего пальца руки вторым и четверто |
синдрома Эдвардса, являются трахеоэзофагальный |
||
го пальца пятым, гипоплазия ногтей, стопа качал |
свищ, аплазия лучевой кости, пороки сердца, неза |
||
ка, синдактилия II–III на стопах (рис. 8). Из поро |
вершенный поворот кишечника. Трисомии как по |
||
ков внутренних органов наиболее часто встречают |
хромосоме 13, так и по хромосоме 18 относятся к |
||
ся пороки развития головного мозга (голопрозэн |
полулетальным синдромам с множественными по |
||
цефалия, аплазия мозолистого тела и др.), пороки |
роками развития. При подтвержденном цитогене |
||
сердца, почек и желудочно кишечного тракта. По |
тическом диагнозе в случае согласия родителей в |
||
роки сердца выявляются более чем у 90% детей с |
большинстве клиник отказываются от проведения |
||
синдромом Эдвардса. Наиболее характерными яв |
хирургической коррекции пороков развития, обес |
||
ляются ДМЖП, ДМПП, ДВПЖ (22, 27). Также |
печивая симптоматическое лечение (31). |
|
|
часто встречаются пупочные грыжи, атрезии пи |
Синдром Шерешевского–Тернера. Пороки |
||
щевода с трахеоэзофагальным свищом, общая |
сердца встречаются и при нарушении количества |
||
1 |
2 |
3 |
|
|
4 |
|
5 |
|
6 |
7 |
8 |
9 |
10 |
11 |
|
12 |
|
13 |
14 |
15 |
|
16 |
17 |
|
18 |
|
|
|
|
|
|
|
|
|
Рис. 7. Кариотип пациента с трисомией 18. |
19 |
20 |
|
21 |
22 |
|
Х |
Y |
Стрелка указывает на дополнительную |
|
|
хромосому 18 |
||||||
|
|
|
|
|
|
|
|
аб
Рис. 8. Ребенок с синдромом Эдвардса: а — положение кисти с перекрывающимися пальцами, характерное для синдрома Эдвардса; б— лицо ребенка с синдромом Эдвардса
110 |
Часть 1. ОБЩИЕ СВЕДЕНИЯ О ВРОЖДЕННЫХ ПОРОКАХ СЕРДЦА |
|
|
|
|
|
|
|
половых хромосом. Врожденные пороки сердца |
фантилизм, аменорея, низкий рост. В большин |
|
присутствуют более чем у 1/3 пациенток с синдро |
стве случаев диагноз ставится именно в пубертат |
|
мом Шерешевского–Тернера (моносомия по Х |
ный период (25). Следует отметить, что деформа |
|
хромосоме, кариотип 45, Х) (рис. 9). Наиболее ти |
ция грудной клетки, которая часто при наличии |
|
пичными являются коарктация аорты, двуствор |
порока сердца оценивается в качестве вторичного |
|
чатый аортальный клапан, атрезия клапана аорты |
изменения, также является одним из характерных |
|
(12). Для периода новорожденности характерны |
признаков синдрома. На 2 м десятилетии жизни |
|
следующие признаки: малая масса при рождении, |
возможно развитие артериальной гипертензии (до |
|
лимфатический отек тыльной поверхности кистей |
30% пациентов). Планируя тактику хирургическо |
|
и стоп (присутствует у 1/3 пациентов в первые ме |
го лечения ВПС, следует учитывать, что при синд |
|
сяцы жизни) (рис. 10), гипоплазия ногтей (обна |
роме Шершевского–Тернера в ряде случаев опи |
|
руживается у 70% пациентов) (Committee on |
сано развитие рекоарктации аорты через несколь |
|
Genetics, Pediatrics, 1995), крыловидные складки |
ко лет после проведенной операции. |
|
на шее (25% больных) (National Institutes of Health, |
Синдромом Вольфа–Хиршхорна. Врожденные |
|
2002), широкая грудная клетка, иногда вдавлен |
пороки сердца имеют место также более чем у 33% |
|
ная, с широко расставленными гипоплазирован |
пациентов с синдромом Вольфа–Хиршхорна |
|
ными сосками (80%) (9). С возрастом задержка |
(синдром делеции короткого плеча хромосомы 4) |
|
роста становится все более заметной, однако в ря |
и являются основной причиной гибели таких па |
|
де случаев врачи связывают ее с наличием у паци |
циентов (5). Характерные признаки этого синдро |
|
ента ВПС. В период полового созревания основ |
ма: лицо в виде греческого шлема с выступающи |
|
ными диагностическими признаками синдрома |
ми лобными буграми, выступающее надпере |
|
Шершевского–Тернера являются: половой ин |
носье, гипертелоризм, гипоплазия надбровных |
|
1 |
2 |
3 |
|
|
4 |
5 |
|
|
6 |
7 |
8 |
9 |
10 |
11 |
12 |
|
|
13 |
14 |
15 |
|
16 |
17 |
18 |
|
|
|
|
|
|
|
|
|
|
Рис. 9. Кариотип при синдроме Шереше |
19 |
20 |
|
21 |
22 |
|
X |
Y |
вского–Тернера. Стрелка указывает на |
|
|
отсутствие 2 й хромосомы Х |
а |
б |
|
|
Рис. 10. Лимфатический отек кистей (а) и стоп (б) у новорожденной с синдромом Шерешевского–Тернера

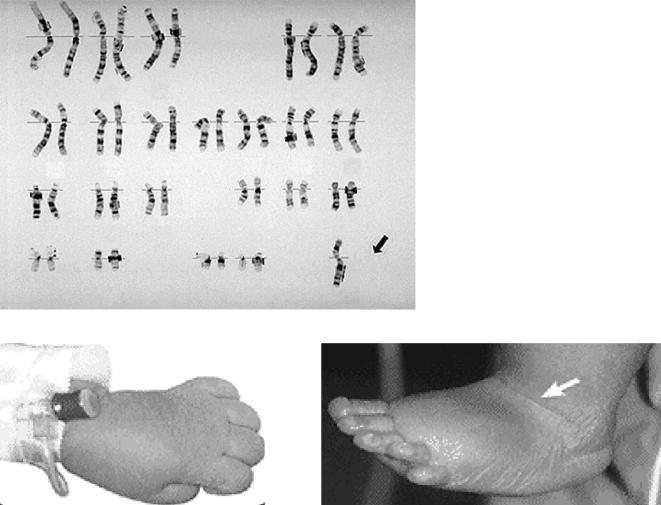
Глава 7. ГЕНЕТИЧЕСКИЕ АСПЕКТЫ ВРОЖДЕННЫХ ПОРОКОВ СЕРДЦА |
111 |
||
|
|
|
|
|
|
|
|
дуг, эпикант, широкий клювовидный нос. Также |
Из этой группы синдромов необходимо выде |
||
встречаются пороки наружных половых органов и |
лить синдром микроделеции 22q11.2 (синдром |
||
у 15% больных — несращения губы и/или нёба |
микроделеции по длинному плечу хромосомы 22) |
||
(29). Кроме того, у подавляющего большинства та |
и синдром Вильямса. |
|
|
ких больных наблюдаются грубая задержка разви |
Синдром микроделеции 22q11.2, или CATCH22 |
||
тия и судороги (4). Врожденные пороки сердца |
синдром (Cardiac defects — пороки сердца, |
||
часто комбинированные, включают пороки разви |
Abnormal faces — лицевой дизморфизм, Thymic |
||
тия клапанов, персистирование левой верхней по |
hypoplasia — гипоплазия тимуса, Cleft palate — рас |
||
лой вены, а также ДМЖП, ДМПП, ОАП, стеноз |
щелина неба, Hypocalcaemia — гипокальциемия |
||
легочной артерии. Для синдрома Вольфа–Хирш |
вследствие делеции |
22q11.2), встречается у 1 из |
|
хорна (рис. 11) характерны также пороки развития |
4000 живых новорожденных (8) и является вторым |
||
головного мозга (гидроцефалия, гипоплазия и ап |
хромосомным синдромом после синдрома Дауна, |
||
лазия мозолистого тела, гипоплазия мозжечка, |
для которого пороки сердца служат диагностичес |
||
зрительного тракта) и почек (поликистоз, гидро |
ким признаком (16). Наиболее характерными яв |
||
нефроз, рефлюксные аномалии, агенезия почки). |
ляются конотрункальные пороки сердца (пороки |
||
Хотя ВПС в большинстве случаев поддаются кор |
выводного тракта) — прерванная дуга аорты, арте |
||
рекции, отдаленный прогноз для жизни часто |
риальный ствол, атрезия легочной артерии. Также |
||
сомнительный, более трети пациентов умирают в |
часто встречаются тетрада Фалло, ДМЖП, аберра |
||
возрасте до 1 года (28). Тяжелое течение послеопе |
нтная правая подключичная артерия (20, 21). Так, |
||
рационного периода объясняется также наличием |
почти 50% пациентов с ОАС имеется данная хро |
||
МВПР и неврологическими нарушениями. |
мосомная аномалия (10), т.е. выявление у пациен |
||
В последние годы в связи с развитием молеку |
та этого порока сердца требует проведения моле |
||
лярно цитогенетических методов диагностики |
кулярно цитогенетического исследования, неза |
||
(fluorescent in situ hybridization — FISH) в отдель |
висимо от наличия или отсутствия других симпто |
||
ную группу выделили так называемые микро |
мов. Другими характерными признаками являют |
||
структурные аномалии хромосом. К этой группе |
ся: малые лицевые аномалии (гипертелоризм, |
||
относятся аномалии строения хромосом, которые |
низкое расположение ушных раковин, круглые |
||
не выявляются при рутинном цитогенетическом |
ушные раковины, широкая переносица), несра |
||
исследовании вследствие своих малых размеров. |
щение нёба, гипокальциемия, гипоплазия тимуса |
||
Для их обнаружения требуется проведение моле |
с нарушением клеточного иммунитета (рис. 12, |
||
кулярно цитогенетических исследований с ис |
13). Следует отметить, что гипокальциемия прису |
||
пользованием специфических ДНК зондов. |
тствует чаще только на 1 м году жизни и носит ла |
||
а |
б |
Рис. 11. Кариотип при синдроме Вольфа–Хиршхорна. Стрелка указывает на делетированную хромосому 4 (а); лицо ребенка с синдромом Вольфа–Хиршхорна (б)