

Неорганическая химия / Астахов М.В. Теория химической связи
.pdf
|
(− |
h2 |
2 |
− |
|
e2 |
)Ψ (2) = E Ψ (2) |
|
|
|||||||||||||||||
|
2m |
|
rb |
|
|
|||||||||||||||||||||
|
|
|
2 |
|
|
|
|
|
|
|
|
b |
|
|
|
|
|
|
0 |
b |
|
(112) |
||||
|
|
|
|
|
|
|
|
|
2 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||
где Е0 — энергия основного состояния водородного атома (EH). |
|
|
||||||||||||||||||||||||
Поэтому интеграл Н11 может быть упрощен |
|
|
|
|
|
|
|
|
|
|
||||||||||||||||
H = |
Ψ |
(1)Ψ |
(2) 2E |
|
|
− |
e2 |
|
− |
|
e2 |
|
+ |
e2 |
|
+ |
e2 |
Ψ (1)Ψ (2)dτ dτ |
|
|||||||
|
|
rb |
|
|
ra |
r12 |
|
|
||||||||||||||||||
11 |
∫∫ |
a |
b |
|
0 |
|
|
|
|
|
|
|
R |
a |
b |
1 2 |
(113) |
|||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||
|
|
|
|
|
|
|
|
1 |
2 |
|
|
|
|
|
|
|
|
|
|
|
||||||
Так как волновая функция 1s нормирована, то |
|
|
|
|
|
|
|
|||||||||||||||||||
|
|
|
|
H =2E + |
e2 |
+I −2I |
2 |
|
|
(114) |
||||||||||||||||
|
|
|
|
|
|
|
||||||||||||||||||||
|
|
|
|
11 |
|
|
|
|
0 |
|
|
|
R |
1 |
|
|
|
|
|
|||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||
где |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
I1 =e2 ∫∫ |
e2 |
|
Ψa(1)Ψb(2)Ψa(1)Ψb(2)dτ1dτ2 |
|
(115) |
||||||||||||||||||
|
|
|
r |
|
||||||||||||||||||||||
|
|
|
|
|
|
12 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
И вследствие равноценности обоих электронов |
|
|
|
|
|
|
|
|||||||||||||||||||
|
|
|
I1 =e2 ∫∫ |
e2 |
|
|
Ψa(1)Ψb(2)Ψa(1)Ψb(2)dτ1dτ2 |
|
(116) |
|||||||||||||||||
|
|
|
r |
|
|
|
||||||||||||||||||||
|
|
|
|
|
|
a |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
2 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
I2 =e2 ∫∫er2 |
|
|
Ψa(1)Ψb(2)Ψa(1)Ψb(2)dτ1dτ2 |
|
(117) |
||||||||||||||||||
|
|
|
|
|
|
b |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
1 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Вычисление этих интегралов еще сложнее, чем соответствующих интегралов для молекулярного иона водорода, и здесь не приводится. Однако и в этом случае может оказаться полезным рассмотрение основной формы выражения энергии.
С помощью тех же доказательств, какие были использованы для H11, можно – показать,
что
|
|
|
|
|
|
|
e2 |
|
|
|
H |
12 |
= 2E S + |
|
+ K − 2K |
2 |
(118) |
||||
|
||||||||||
|
|
|
|
0 |
12 |
1 |
|
|||
|
|
|
|
|
|
|
rab |
|
|
|
где |
|
|
|
|
|
|
|
|
|
|
K1 =e2 ∫∫ |
e2 |
Ψa(1)Ψb(2)Ψa(2)Ψb(1)dτ1dτ2 |
(119) |
|||||||
r |
||||||||||
|
|
12 |
|
|
|
|
|
|
||
и вследствие равноценности электронов |
|
|
|
|
||||||
K2 =e2 ∫∫er2 |
[Ψa(1)Ψb(2)Ψa(2)Ψb(1)]dτ1dτ2 |
(120) |
||||||||
|
|
|
|
a |
|
|
|
|
|
|
|
|
1 |
|
|
|
|
|
|
||
K2 =e2 ∫∫ |
e2 |
[Ψa(1)Ψb(2)Ψa(2)Ψb(1)]dτ1dτ2 |
(121) |
|||||||
r |
||||||||||
|
|
|
|
b |
|
|
|
|
|
|
|
|
2 |
|
|
|
|
|
|
||
41
Если эти результаты подставить в выражения для энергии молекулы водорода, то после преобразования получим уравнения, соответствующие симметричному и антисимметричному состояниям:
E |
− 2E |
= |
e2 |
+ |
I − 2I |
2 |
+ K − 2K |
2 |
|
(122) |
|||||||
|
|
1 |
|
1 |
|
||||||||||||
|
|
|
|
|
|
|
|
||||||||||
|
c |
0 |
rab |
|
|
|
|
1+ S12 |
|
|
|
|
|||||
|
|
|
|
|
|
|
|
|
|
|
|
||||||
E |
|
− 2E |
= |
|
e2 |
|
+ |
|
I − 2I |
2 |
− K + 2K |
2 |
(123) |
||||
ac |
|
|
|
|
1 |
|
|
1 |
|
||||||||
|
|
|
|
|
|
|
|
|
|
||||||||
|
0 |
|
rab |
|
|
|
|
|
1− S12 |
|
|
|
|
||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||
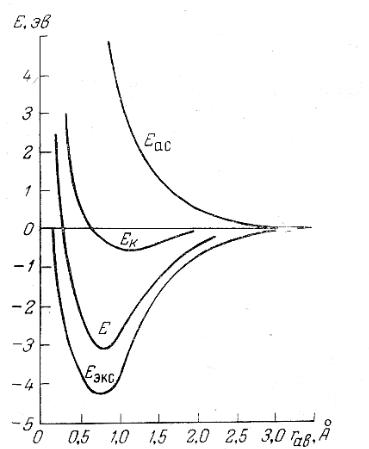
Кривые потенциальной энергии, получающиеся для симметричного и антисимметричного состояний, приведены на рис. 14.
Рис. 14 Кривые потенциальной энергии молекулы водорода.
Еас - для антисимметричного состояния; Ек - для классического кулоновского взаимодействия; Е – расчетное значение энергии связи; Еэк – экспериментальное значение.
Если принять энергию изолированного атома водорода в основном состоянии за нуль, т. Е. Е0 = 0, то получившиеся кривые потенциальной энергии показывают энергию взаимодействия между двумя атомами водорода, когда они образуют молекулу. Кривая антисимметричного состояния не имеет минимума и характеризует неустойчивое состояние.
С другой стороны, на кривой симметричного состояния есть довольно глубокий минимум, указывающий на устойчивость молекулы. Замечательно, что при этой начальной функции минимум потенциальной энергии значительно ближе к экспериментальному, чем мини-
42
мумы, полученные без учета неразличимости электронов. Однако надо сказать, что, используя другие волновые функции, можно получить еще лучшее совпадение результатов расчета и опыта.
Признание неразличимости электронов в молекуле водорода равноценно признанию существования двух структур
На(1);Нb(2) и На(2)Нb(1)
I II
Так как оба изолированных атома водорода одинаковы, то и волновые функции и энергии структур I и II одинаковы. Чтобы оценить значение неразличимости обоих электронов, необходимо рассмотреть энергию молекулы с помощью одной или другой из этих структур.
Если, например, принять структуру I, то можно написать соответствующую волновую функцию
|
|
Ψ1=ψa(1) ψ2(2) |
(124) |
||
|
|
Ψ1=ψa(1) ψ2(2) |
(124) |
||
и энергия системы будет |
|
|
|
||
E = |
∫ΨHΨdτ |
= |
∫ψa (1)ψb (2)Hψa (1)ψb (2)dτ |
(125) |
|
∫ΨΨdτ |
∫ψa (1)ψb (2)ψa (1)ψb (2)dτ |
||||
|
|
|
|||
или просто |
|
|
|
||
|
|
|
Ek = H11 |
(126) |
|
|
|
|
S11 |
|
|
Это выражение можно еще упростить, используя нормированную 1s-волновую функцию, тогда S11 = 1 и
Eк = H11 |
(127) |
В соответствии с уравнением (114), можно написать, что
|
|
|
e2 |
|
|
|
E |
k |
= 2E + |
|
+ I − I |
2 |
(128) |
|
||||||
|
0 |
1 |
|
|||
|
|
|
rab |
|
|
|
Энергия взаимодействия между двумя атомами может быть определена, если приравнять энергию изолированных атомов E0 нулю. Получившаяся кривая потенциальной энергии показывает уменьшение энергии системы, по сравнению с энергией изолированных атомов, вследствие сближения атомов водорода. Если на кривой потенциальной энергии имеется минимум, значит, теория предсказывает существование устойчивой молекулы. На рис. 14 вторая кривая изображает Eк, на ней виден небольшой минимум, что указывает на устойчивость молекулы, но вычисленная величина энергии составляет всего около 0,1 от экспериментального значения.
Если разобрать отдельные члены выражения для энергии, можно сделать вывод, что Ек по существу является энергией классического кулоновского взаимодействия между двумя атомами водорода. Интеграл I1 представляет собой величину
43

I1 = e2 ∫∫ |
1 |
ψa2 (1)ψb2 (2)dτ2dτ1 |
(129) |
r |
|||
12 |
|
|
|
где e2 энергия электростатического отталкивания между двумя зарядами;
r12
ψa2(1) ψb2(2) — плотности заряда электронов (I) и (2).
Можно сделать заключение, что I1 характеризует электростатическое отталкивание между электронными облаками (1) и (2). С другой - стороны, интеграл I2 имеет вид
I2 =e2 ∫∫ |
e2 |
[Ψa (1)Ψb (2)Ψa (1)Ψb (2)]dτ1dτ2 =e2 ∫∫ |
e2 |
[Ψa (1)Ψb (2)Ψa (1)Ψb (2)]dτ1dτ2 |
(130) |
r |
r |
||||
|
a2 |
|
b1 |
|
|
Его можно преобразовать
I2 = e2 ∫ |
1 |
ψb2 (2)dτ2 |
= e2 ∫ |
1 |
ψa2 (1)dτ1 |
(131) |
|
r |
r |
||||||
|
a |
2 |
|
|
b |
|
|
|
|
|
1 |
|
|
||
Это выражение можно трактовать как притяжение между электроном одного атома и ядром другого атома, например, между положительным зарядом ядра b и отрицательным зарядом электронного облака вокруг ядра а.
Наконец, e2/rab выражает отталкивание между двумя положительно заряженными ядрами. Таким образом, имеются два члена, обозначающих электростатическое отталкивание
— e2/rab и I1 один член электростатического притяжения — 2I2.
Величина Ек выражает классическое электростатическое притяжение между двумя атомами водорода, которое приводит к образованию устойчивой молекулы водорода, но вычисленная устойчивость этой молекулы не так велика, как показывает опыт.
Признание возможности существования двух структур I и II обусловливает новую волновую функцию. Энергия диссоциации, вычисленная с использованием этой новой функции, составляет уже 70% от экспериментальной величины. Если иметь в виду, что энергия диссоциации, вычисленная на основе простого электростатического взаимодействия, равна всего 10% от экспериментальной величины, то можно понять, почему новый результат рассматривают как достижение квантовомеханического метода. На языке теории валентных связей этот эффект называют резонансом, и он играет решающую роль в этой теории. Можно сделать заключение, что электрон (1) в данный момент находится около атома а, а электрон (2) около атома b, а в следующий момент они находятся в противоположном положении. Таким образом, электроны находятся в резонансе между двумя структурами. Однако в настоящее время такая точка зрения не принята. Более правильно считать, что истинное состояние системы не соответствует ни структуре I, ни структуре II, но представляет собой нечто среднее, имеющее характер каждой из независимых структур. Так строят волновую функцию для описания истинной структуры, вводя общие черты, характерные для каждой из индивидуальных структур. Результат, полученный с помощью этой новой функции, описывает более устойчивое состояние, чем с помощью функций, описывающих каждую отдельную структуру; и разность соответствующих энергий называется энергией резонанса. Важно отдавать себе отчет в том, что резонанс в некотором смысле фиктивное понятие. Оно возникает как следствие построения начальной волновой функции, и его существование есть продукт квантовомеханической модели, использованной для описания системы.
44
Поскольку применяют теорию валентных связей, постольку существует и представление о резонансе, так как оно лежит в основе этой теории. Однако для другой, не менее хорошей модели оно не будет иметь смысла, а, следовательно, не будет иметь право на существование. То же самое относится, разумеется, и к некоторым другим научным концепциям. В теории валентных связей резонанс существует, и можно считать себя вправе говорить о нем как о существующем. Но в свете других моделей можно отрицать его существование.
Исходную волновую функцию можно улучшить, допустив, что к общей волновой функции водородной молекулы прибавляются еще функции двух дополнительных структур. Это ионные структуры:
:Ha- Hb+ Ha+ :Hb-
IIIIV
вкоторых оба электрона расположены или около атома а, или около атома b. Очевидно, эти структуры не могут иметь равное значение с гомеополярными структурами I и II. Следовательно, полная волновая функция может быть выражена так
Ψвс=aΨ1+aΨII+bΨIII+bΨIV=aψa(1)ψb(2)+aψa(2)ψb(1)+ bψa(1)ψa(2) + bψb(1)ψb(2) (132)
Как видно, оба гомеополярных члена имеют одинаковое значение, как и оба ионных члена. В общем случае можно считать, что
Ψвс =aΨ1 + bΨII +cΨIII +dΨIV+ |
(133) |
Значения параметров а, b, с, d и т. д. могут быть найдены посредством минимизации энергии системы по каждому параметру. Подобный расчет для молекулы водорода показывает, что связь имеет на 17% ионный характер. Надо заметить, что по теории молекулярных орбиталей и простая волновая функция для водородной молекулы содержит ионные члены, соответствующие структурам III и IV, но им придан такой же вес, как и гомеополярным структурам.
3.3Насыщенность и направленность ковалентной связи.
Одним из наиболее выдающихся достижений квантовомеханического подхода к строению молекулы был его успех в области молекулярной геометрии. Рассматривая связи между атомами в молекуле как результат перекрывания атомных орбиталей, следует ожидать от молекулы определенной пространственной структуры.
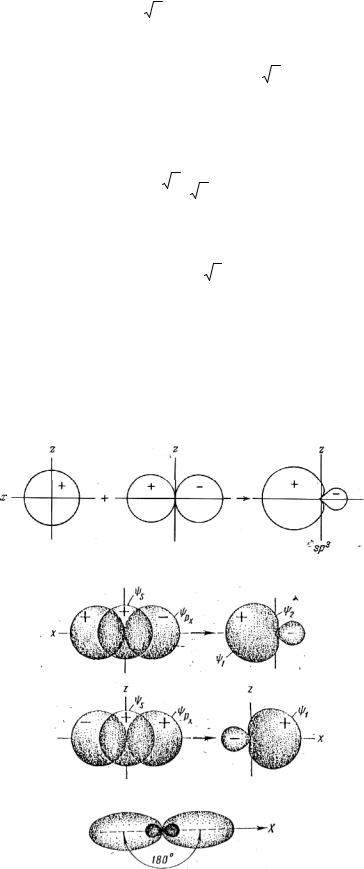
Существует много различных путей объяснения строения молекулы. Стереохимический метод отличается от излагаемого здесь, но использование понятия локализованных атомных орбиталей в теории валентных связей оказалось настолько удачным, что есть смысл это обсудить. Согласно теории валентных связей, ковалентную связь можно представить как результат спаривания двух электронов на атомных орбиталях двух разных атомов. Затем связь должна расположиться в направлении перекрывания атомных орбиталей. Наиболее прочная связь образуется в том месте, где возможно наибольшее перекрывание двух орбиталей. В качестве примера рассмотрим молекулу воды. Связь двух водородных атомов образуется орбиталями 1s. У кислородного атома электронная конфигурация 1s22s22px22py12pz1, и так как считается, что связь образуется посредством спаривания электронов, то, по-видимому, будут спариваться 2py1 и 2pz1-электроны. На рис. 15 показана yz
45

плоскость молекулы воды, орбитали ру- и рz-расположены в этой плоскости взаимно перпендикулярно. Для получения возможно большего перекрывания между 1s-орбиталями атомов водорода и ру- и рz-орбиталями атома кислорода необходимо, чтобы атомы водорода подошли к атомам кислорода вдоль осей y и z. Это дает показанное на рисунке перекрывание, а угол НОН должен быть равен примерно 90°. На самом деле этот угол равен 104°31’; такое отклонение от 90° может быть вызвано отталкиванием между двумя атомами водорода, а также некоторым участием в связи 2s-электронов атома кислорода.
Рис. 15. Перекрывание связывающих орбиталей в молекуле воды.
Далее будет видно, что связь можно рассматривать как гибридную sp2-связь. Такую же структуру, как у воды, можно ожидать у Н2S, Н2Sе и Н2Те. Действительно, сходство между теорией и опытом для этих молекул вполне хорошее: угол НХН для Н2S найден 92,2°, для Н2Sе — 91,0° и для Н2Те —89,5°.
Такие же общие рассуждения можно применить к ряду других соединений: NН3, РН3, АsН3 и SbН3. В этом случае в образовании связей участвуют все три р-орбитали и углы НХН должны быть также 90°. На опыте углы соответственно равны 107,3°, 93,3°, 91,8° и 91,3°. Отклонение от теории можно объяснить как с точки зрения теории валентных связей, так и с помощью различных теоретических моделей. По этим простым схемам видно, что представление о перекрывании атомных орбиталей является основой для молекулярной геометрии.
3.4 Гибридизация
Хотя простое объяснение, основанное на перекрывании s- и р-орбиталей, вполне подходит для многих молекул, оно совершенно неприменимо для соединений углерода. В атоме углерода заселенность атомных орбиталей следующая:
↑↓ ↓↑ ↓ ↓
1s 2s 2px 2py
На основании такой конфигурации можно ожидать образования молекулы, подобной воде, в которой атом углерода имел бы валентность 2 с взаимно перпендикулярными связями. Этого у атома углерода не наблюдается, его четырехвалентность и тетраэдрическая структура хорошо известны.
Четырехвалентность углерода можно объяснить тем, что один из 2s-электронов возбуждается при переходе на пустую 2рz-орбиталь с образованием конфигурации
46
↑↓ ↓ ↓ ↓ ↓
1s 2s 2px 2py 2pz
Согласно теории валентных связей, такая конфигурация предсказывает, что в молекуле, например СН4, три атома водорода энергетически эквивалентны и их связи взаимно перпендикулярны, а четвертый атом водорода удерживается более слабой связью и находится под углом 125° по отношению к остальным связям, Однако в действительности все четыре связи углерода в органических молекулах эквивалентны, т. е, наблюдается противоречие. Вопреки кажущимся трудностям, задача может быть решена с помощью теории валентных связей. В предыдущем изложении теории валентных связей было показано, что волновая функция улучшается путем линейных комбинаций нескольких разумных функций, описывающих различные представления о молекуле, т. е. ее резонансные структуры. Так же и в данном случае можно предположить, что истинные волновые функции четырех связей углерода должны быть представлены как комбинация функций s- и р-орбиталей, участвующих в образовании связей. Самой лучшей комбинацией будет та, которая соответствует наиболее прочной связи. Однако не совсем ясно, чем измерять силу связи. Кажется разумным принять, что наиболее прочной будет связь, допускающая наибольшее возможное перекрывание между связывающими орбиталями. Это условие называют критерием наибольшего перекрывания, и оно является основой при рассмотрении направленных связей с точки зрения теории валентных связей. При построении гибридных орбиталей важно, чтобы радиальные части орбиталей данного электронного уровня были приблизительно одинаковы. На основании этого допускают, что гибридная орбиталь может быть построена из угловых частей индивидуальных волновых функций. Например, для четырехвалентного атома углерода это будут четыре связи, описываемые волновыми функциями вида
Ψ=aiφs + biφpy + ciφpx + diφpz |
(136) |
где φi— чисто угловые волновые функции.
Так как радиальная часть волновых функций не принимается во внимание, индивидуальные угловые волновые функции теперь могут быть выражены посредством сферических гармоник, как показано в таблице 7.
Таблица 7. Сферические гармоники для s и р-подуровней атома водорода.
|
Подуровни |
Угловые составляющие. |
l- |
m |
0 |
0 |
|
Ф0,0 (ϑ,ϕ) = |
|
|
1 |
|
|||||
|
|
|
4π |
|||||||||
|
|
|
|
|
|
|
|
|
|
|
||
1 |
0 |
Ф (ϑ,ϕ) = |
|
3 |
4 |
π cosϑ |
||||||
|
|
|
1.0 |
|
|
|
|
|
|
|
||
|
|
|
|
|
|
|
|
|
|
|
||
1 |
1 |
Ф (ϑ,ϕ) = |
3 |
4 |
π sinϑcosϕ |
|||||||
|
|
1.1 |
|
|
|
|
|
|
|
|
||
|
|
|
|
|
|
|
|
|
|
|
||
1 |
-1 |
Ф |
(ϑ,ϕ) = |
3 |
4 |
π sinϑsinϕ |
||||||
|
|
1.−1 |
|
|
|
|
|
|
|
|
||
|
|
|
|
|
|
|
|
|
|
|
||
47

Заметим, что Φ = |
1 |
,а все р-орбитали содержат множитель Ф = |
3 |
4 |
π . Ради про- |
|
|||||
|
4π |
|
|
||
|
|
|
|
||
стоты φs, принимают за единицу, а все функции нормируются к 4π. Отсюда получаются уравнения для индивидуальных угловых волновых функций:
φs =1 |
(137а) |
φpx=√3 sinθcosφ |
(1376) |
φpy=√3 sinθsinφ |
(137в) |
φpz=√3 cosθ |
(137г) |
Применяя требование нормирования, можно сказать, что для каждой из этих функций, а также для их линейных комбинаций
∫ φiφi*dτ= 1
или выполняя интегрирование по θ и φ по поверхности сферы
2ππ |
|
|
∫∫ψIψI* sinϑdϑdϕ |
(138) |
|
0 |
0 |
|
Вследствие ортогональности различных атомных орбиталей на коэффициенты в уравнении (136) накладывается ограничение
ai+bi+ci+di= 1 |
(139) |
Для определения угловых соотношений между связями удобно принять направление первой связи вдоль одной из осей, например оси х. При этом условии орбитали рy и рz не будут участвовать в этой связи, поэтому ci и di будут равны нулю:
Ψ1=a1 ψs+b1ψpx |
(140) |
|
Так как аi + bi = 1 , то |
|
|
Ψ = a + |
1− a2ψ |
(141) |
1 1 |
1 |
px |
Если в последнее уравнение подставить значения волновых функций при условии ψs=1, то получим
Ψ = a + |
1− a2ψ |
(142) |
1 1 |
1 |
px |
Применяя принцип наибольшего перекрывания при условии наиболее прочной связи, надо выбрать параметр a1 так, чтобы функция Ψ1 была как можно большей в направлении связи. Это основано на предположении, что чем больше протяженность орбитали в пространстве, тем более прочную связь она может образовать. Величина Ψ1вдоль оси х равна
Ψ = a + |
3 × 1− a2ψ |
(143) |
1 1 |
1 |
px |
так как произведение sinθ соsφ равно единице при θ = π/2 и φ= 0. Это и будет наибольшей возможной величиной для данного члена. Если последнее уравнение продифференцировать по а1 и приравнять результат нулю, получится
48
|
1− a |
|
3(1− a2 )−1/ 2 = 0 |
(144) |
|||||||||||||||
|
|
|
|
|
1 |
|
|
|
|
|
1 |
|
|
|
|
|
|||
откуда |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
a = |
1 |
|
|
|
|
|
|
|
|
и |
|
|
b |
= |
3 |
|
|||
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||
1 |
|
2 |
|
|
|
|
|
|
|
|
|
|
|
|
1 |
2 |
|
||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||
Тогда значение волновой функции для первой гибридной связи [из ур. (142)] будет |
|||||||||||||||||||
|
|
|
|
|
1 |
|
|
|
|
|
3 |
|
|
|
|
|
|
|
|
Ψ = |
|
|
|
+ |
|
|
|
|
|
3 sinϑcosϕ |
(145) |
||||||||
|
|
|
|
|
|
|
|||||||||||||
|
1 |
|
|
|
2 |
|
|
|
2 |
|
|
|
|
|
|
|
|
||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||
а в орбитальных символах |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
1 |
|
|
|
|
|
3 |
|
|
|
|
|
|
|
Ψ = |
ψ |
|
+ |
|
|
|
|
(146) |
|||||||||
|
|
|
|
|
|
|
|
||||||||||||
|
|
|
s |
|
|
|
ψ |
px |
|||||||||||
|
|
1 |
|
|
2 |
|
|
|
2 |
|
|
||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||
Выше ψs была приравнена единице, и на этом основании р-орбитали приняли выражение, указанное в уравнениях (137). Если положить sinθ и cosφ равными единице и тем самым оценить наибольшее значение в пространстве р-орбитали, то получится величина 1,732. Это показывает, что р-орбиталь способна к более сильному перекрыванию, чем чистая s- орбиталь, и, следовательно, pх
Рис. 16. Образование и структура sр-гибридных орбиталей
49
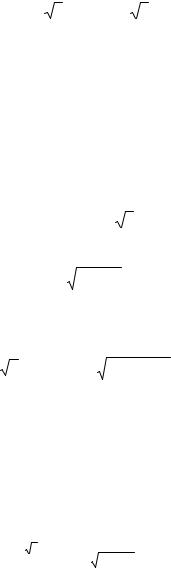
должна давать более прочную связь. На том же основании можно определить величины гибридной связи Ψ1. Считая sinθ и cosφ равными единице, для Ψ1 в направлении связи
.получим величину 2. Это значительно больше, чем для чистых s- и р-орбиталей, и поэтому можно ожидать, что гибридные связи будут более прочными, чем чистые s- или р-связи.
Образование гибридной связи показано графически на рис. 16. s-oрбиталь имеет только положительный знак, а р-орбиталь имеет положительную и отрицательную части. Положительная часть комбинируется с s-орбиталью, а отрицательная уменьшается вследствие слияния.
Для второй гибридной связи можно выбрать плоскость хz. В этом случае не будет участвовать орбиталь ру, и новая волновая функция будет
Ψ2=a2φs + b2φpy + c2φpx + d2φpz |
(147) |
Если орбиталь лежит в плоскости хz, угол φ будет равен 0° или 180°. Углу φ = 0 соответствует область Ψ1, поэтому Ψ2 будет иметь максимум при φ = 180°, тогда cosφ будет равен -1.
Учитывая это и подставляя выражение для волновой функции в уравнение (147), полу-
чим
Ψ2 = a2 +b2 3 sinϑ + d2 3 cosϑ |
(148) |
Это уравнение можно выразить через параметр b2 и угол d2, используя соотношения
a22+ b22+ d22=1 |
(149) |
a1a2 +b1b2=0 |
(150) |
Второе уравнение получается из условия ортогональности. Так как d2 равно нулю, то в последнем соотношении отсутствует произведение d1d2 . Зная величины а1 и b1, можно выразить a2 и d2 через b2:
|
|
|
|
|
|
a2 |
|
b1 |
|
= − 3b2 |
(151) |
|
|||||
= − a |
b2 |
||||
|
1 |
|
|
|
|
d |
2 |
= ± 1− 4b2 |
(152) |
|
2 |
|
Какой знак выбрать для d2 несущественно, поэтому ради удобства принимают положительный знак. Теперь можно выразить Ψ2
Ψ |
= −b |
3(1+sinϑ) + 3(1− 4b2 ) cosϑ |
(153) |
2 |
2 |
2 |
|
Так же как и для первой гибридной орбита ли, чтобы определить параметр b2, необходимо найти максимальное значение Ψ2 в направлении связи. Однако в этом случае переменным является также угол θ, он и определяет направление связи. Следовательно, надо искать максимум функций как в отношении b2, так и θ. Это приводит к двум уравнениям, которые могут быть решены одновременно. Первое из них получается путем дифференцирования Ψ2 по b2 и приравниванием производной нулю
dΨ2 |
|
|
12b2 |
|
(154) |
|
|
|
|
= − 3(1+sinθ) + |
|
cosθ = 0 |
|
db |
|
2 |
|
|||
|
2 |
θ |
|
3 −12b2 |
|
|
50